Voltammetric determination of hydroxylamine at the surface of a quinizarine/TiO2 nanoparticles-modified carbon paste electrode
M.
Mazloum-Ardakani
*a,
H.
Beitollahi
a,
Z.
Taleat
a and
H.
Naeimi
b
aDepartment of Chemistry, Faculty of Science, Yazd University, Yazd, 89195-741, I.R. Iran. E-mail: mazloum@yazduni.ac.ir; Fax: +00983518210644; Tel: +00983518211670
bDepartment of Organic Chemistry, Faculty of Chemistry, University of Kashan, Kashan, I.R. Iran
First published on 14th September 2010
Abstract
A carbon-paste electrode (CPE) was chemically modified with TiO2 nanoparticles and quinizarine (QZ). This was used as an electrochemical sensor for the determination of minor amounts of hydroxylamine (HX). The modified electrode showed very efficient electrocatalytic activity for the anodic oxidation of hydroxylamine, owing to a substantial decrease in anodic overpotentials. Under optimal conditions, the linear range span of HX concentration was 1.0 μM to 400.0 μM and the detection limit was 0.173 μM at a signal-to-noise ratio of 3, using differential pulse voltammetry (DPV). The diffusion coefficient and kinetic parameters (e.g., electron transfer coefficient for HX) were also determined using electrochemical approaches. The sensor demonstrated good stability and reproducibility. To evaluate the applicability of the proposed method to real samples, the modified CPE was applied to the determination of HX in water samples.
1. Introduction
Hydroxylamine (HX) is known as a kind of reducing agent, and is widely used for industrial and pharmaceutical applications. It has been identified as a key intermediate in nitrogen cycles and nitrous oxide production.1 HX is a well-known mutagen, and is moderately toxic and harmful to humans, animals, and even plants.2 It is widely used as a raw material for the synthesis of pharmaceutical intermediates and final drug substances. Thus, the quantitative determination of HX is very important for biological process studies and industrial purposes.3 Many methods have been developed for the determination of HX. For example, chromatographic,4 spectrophotometric,5–7 and electrochemical detection (ECD)8–17 methods have been successfully applied toward HX determination.Electrochemical analysis is gaining in importance within industrial process control, environmental monitoring, and various medicine and biotechnology applications.18–21 The use of bare electrodes for electrochemical detection has a number of limitations, such as low sensitivity, poor reproducibility, slow electron transfer reaction, low stability over a wide range of solution compositions, and high overpotential at which the electron transfer process occurs.22 Chemical modification of inert substrate electrodes with redox active thin films offers significant advantages in the design and development of electrochemical sensors. In operation, the redox active sites shuttle electrons between the analyte and the electrodes with a significant reduction in activation overpotential.23 A further advantage of chemically modified electrodes is that they are less prone to surface fouling and oxide formation compared to inert substrate electrodes.24 A wide variety of compounds have been used as electron transfer mediators for the modification of electrode surfaces with various procedures.25–31
New input into the field of electroanalysis and especially bioelectroanalysis came with rapid developments in nanotechnology at the break of the 21st century. The unique chemical and physical properties of nanoparticles make them extremely suitable for designing new and improved sensing devices, especially electrochemical sensors and biosensors.32 Many kinds of nanoparticles, such as metal-oxide and semiconductor33,34 nanoparticles, have been used to construct electrochemical sensors and biosensors. These nanoparticles play different roles in different sensing systems. The important functions provided by nanoparticles include: the immobilization of biomolecules; the catalysis of electrochemical reactions; the enhancement of electron transfer between electrode surfaces and proteins; and the labeling of biomolecules, including their action as reactants.
In the present work, we describe the preparation and suitability of a quinizarine (QZ)-modified TiO2 nanoparticle carbon paste electrode (QZMNCPE) as a new electrode in the electrocatalysis and determination of HX in an aqueous buffer solution. To demonstrate the catalytic ability of the modified electrode toward the electrooxidation of HX in real samples, we examined the utility of this method for the voltammetric determination of HX in water samples.
2. Experimental
2.1. Apparatus and chemicals
Electrochemical experiments were carried out using a computerized potentiostat/galvanostat μAutolab Type III (Eco Chemie B.V.A.). The experiments were controlled with General Purpose Electrochemical System (GPES) software. A conventional three electrode cell was used with Ag/AgCl/KCl (3.0 M concentration), platinum wire, and QZMNCPE as the reference, auxiliary, and working electrodes, respectively. A Metrohm model 691 pH/mV meter was also used for pH measurements.All solutions were freshly prepared with doubly distilled water. The HX was from Fluka and was used as received. Other reagents were analytical grade (Merck). Graphite powder (Merck) and paraffin oil (DC 350, Merck, density = 0.88 g cm−3) were used as binding agents for graphite pastes. Buffer solutions were prepared from orthophosphoric acid, and its salts in the pH range between 2.0 and 11.0. TiO2 nanoparticles (surface area = 84 m2g−1 and particle size = 6.7 nm)35 and QZ31 were synthesized in our laboratory.
2.2. Electrode preparation
Modified electrodes were prepared by the following steps. 0.01 g QZ was dissolved in CH3Cl. This was hand mixed, using a mortar and pestle, with 95 times its weight in graphite powder and 4 times its weight in TiO2 nanoparticles. Paraffin was added to the mixture and mixed for 20 min until a uniformly wetted paste was obtained. This paste was then packed into the end of a glass tube (ca. 3.4 mm i.d. and 10 cm long). A copper wire inserted into the carbon paste provided an electrical contact. When necessary, a new surface was obtained by pushing excess paste out of the tube, which was then polished with weighing paper.Unmodified carbon paste was prepared in the same way without adding QZ and TiO2 nanoparticles to the mixture. This was used for the purpose of comparison.
3. Results and discussion
3.1. Electrochemistry of QZMNCPE
QZ is insoluble in aqueous solutions. It can be easily incorporated into the carbon paste without its leaching from the electrode surface. Thus, this material can be fabricated into a reproducible chemically modified electrode.The electrochemical behavior of the QZMNCPE was first investigated using cyclic voltammetry. Experimental results show well-defined and reproducible anodic and cathodic peaks with Epa = 0.560 V, Epc = 0.400 V, E0′ = 0.480 V vs. Ag/AgCl/KCl 3.0 M and ΔEp = 160 mV. The electrode process was quasi-reversible, with ΔEp greater than the (59/n) mV expected for a reversible system.
The capability of the electrode to generate a reproducible surface was examined using cyclic voltammetric data obtained from optimum solutions (pH 7.0) with five separately prepared QZMNCPEs. The calculated RSDs for various parameters were accepted as the criteria for satisfactory surface reproducibility (about 1–4%). This is virtually the same as that expected for renewal on ordinary carbon paste surfaces. However, we regenerated the QZMNCPE surface before each experiment, as per our previous result.31
Cyclic voltammograms of the QZMNCPE were recorded at different scan rates (from 25 to 900 mVs−1). Fig. 1A illustrates that the anodic and cathodic peak currents (Ip) were linearly dependent on ν at scan rates between 25 and 500 mV s−1. A linear correlation was obtained between peak currents and the scan rate, indicating that the redox process was not controlled by diffusion. Fig. 1B shows the anodic peak potentials (Epa) as a function of the potential sweep rate. We found that, for scan rates above 200 mV s−1, the Ep values were proportional to the logarithm of the scan rate. Under these conditions, ks can be obtained according to the following equation:36
| Log(ks) = α log (1 − α) + (1 − α) log α − log (RT/nFν) − α (1 − α) nαFΔEp/2.3RT | (1) |
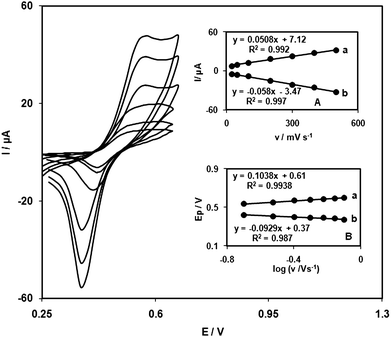 | ||
| Fig. 1 Cyclic voltammograms of QZTNMCPE in 0.1 M phosphate buffer (pH 7.0) at various scan rates (from inner to outer) 25, 50, 100, 200, 300, 400 and 500 mVs−1. (A) Plot of anodic (a) and cathodic (b) peak currents of QZTNMCPE vs. v from cyclic voltammograms. (B) Variation of Epa and Epcversus the logarithm of the high scan rates. | ||
3.2. Electrocatalytic oxidation of HX
Fig. 2 depicts the cyclic voltammetric responses from the electrochemical oxidation of 1 mM HX at: QZMNCPE (curve f), QZ-modified CPE (QZMCPE; curve e), carbon TiO2 nanoparticles (CNPE; curve d), and bare CPE (curve a). As shown, the anodic peak potential for the oxidation of HX at QZMNCPE (curve f) and QZMNCPE (curve e) was about 560 mV, while at the CNPE (curve d) and bare CPE (curve b) HX was not oxidized until the potential reached +1200 mV. These results demonstrate that the best electrocatalytic effect for HX oxidation was observed at QZMNCPE (curve f). Also, when we compared the oxidation of HX at a QZMCPE (curve e) versus a QZMNCPE (curve f), there was a dramatic enhancement of the anodic peak current at QZMNCPE relative to the value obtained at the QZMCPE. In other words, the obtained data clearly show that the combination of TiO2 nanoparticles and a mediator (QZ) definitely improve the characteristics of HX oxidation. The QZMNCPE in 0.1 M phosphate buffer (pH 7.0), without HX in solution, exhibited a well-behaved redox reaction (curve c). Upon the addition of 1 mM HX the anodic peak current of the mediator was greatly increased, while the corresponding cathodic peak disappeared in the reverse scan of the potential (curve f). This behavior is typical of that expected for electrocatalysis at chemically modified electrodes.37 | ||
| Fig. 2 Cyclic voltammograms of (a) CPE in 0.1 M phosphate buffer solution (pH 7.0) at a scan rate of 25 mV s−1 and (b) as (a) 1 mM HX (c) as (a) and (d) as (b) at the surface of QZTNMCPE and TNCPE, respectively. Also, (e) and (f) as (b) at the surface of QZMCPE and QZTNMCPE, respectively. | ||
The effect of scan rate on the electrocatalytic oxidation of 0.75 mM HX at the QZMNCPE was investigated by cyclic voltammetry (Fig. 3). The oxidation peak potential shifted with increasing scan rates towards a more positive potential, confirming the kinetic limitation of the electrochemical reaction. A plot of peak height (Ip) against square root of scan rate (ν1/2), in the range of 5–30 mV s−1, was constructed (Fig. 3A, curve a). This was found to be linear, suggesting that at sufficient overpotential the process is diffusion rather than surface controlled. A plot of the sweep rate normalized current (Ip/ν1/2) versus sweep rate (Fig. 3A, curve b) exhibits the characteristic shape typical of an EC process.37
 | ||
| Fig. 3 Cyclic voltammograms of a QZTNMCPE in 0.1 M phosphate buffer (pH 7.0) containing 0.75 mM HX at different scan rates: (a) 5, (b) 10, (c) 15, (d) 20, (e) 25, and (f) 30 mV s−1. Insets: (A) Curve a, plot of Ivs.ν1/2, and curve b, plot of Iν−1/2vs.ν. (B) Curve a, plot of Epvs. log ν, and curve b, Tafel plot derived from the rising part of the voltammogram recorded at a scan rate of 10 mV s−1. | ||
To obtain information on the rate-determining step, a Tafel slope (b) was determined using the following equation for a totally irreversible diffusion controlled process:38
| Ep = (b/2) log ν + constant | (2) |
Curve b in Fig. 3B shows a Tafel plot that was drawn from data from the rising part of the current–voltage curve, which were recorded at a scan rate of 10 mV s−1. This part of the voltammogram, known as the Tafel region, is affected by electron transfer kinetics between HX and the surface-confined QZ, assuming that deprotonation of the substrate is a sufficiently rapid step. In this condition, the number of electrons involved in the rate-determining step can be estimated from Tafel plot slope. A slope of 0.1002 V decade−1 was obtained, indicating that a one electron transfer was rate limiting, assuming a transfer coefficient of α = 0.41.
3.3. Chronoamperometric studies
The catalytic oxidation of HX by a QZMNCPE was also studied by chronoamperometry. Chronoamperometric measurements using different concentrations of HX at QZMNCPEs were performed by setting the working electrode potential to 700 mV (Fig. 4). In chronoamperometric studies, we have determined the diffusion coefficient (D) of HX. Experimental plots of Iversust−1/2, with the best fits for different HX concentrations, were employed. The slopes of the resulting straight lines were then plotted versus HX concentrations (Fig. 4, inset A). Using the Cottrell equation37 and these slope values, we calculated a diffusion coefficient of 6.1 × 10−5 cm2 s−1 for HX.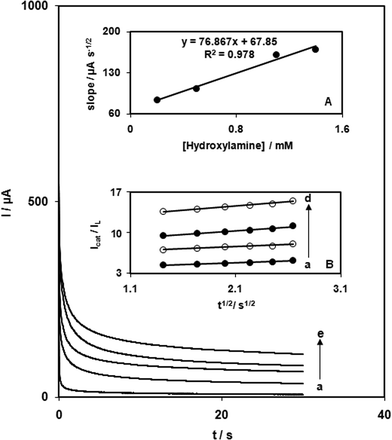 | ||
| Fig. 4 Chronoamperometric response at QZTNMCPE in 0.1 M phosphate buffer (pH 7.0) for different concentrations of HX: (a) 0, (b) 0.2, (c) 0.5, (d) 1.1, and (e) 1.4 mM. Inset: (A) Plot of the slope of straight lines against the HX concentration. (B) Dependence of Icat/Il on t1/2 derived from chronoamperogram data. | ||
Chronoamperometry can also be used to estimate the catalytic rate constant (k) for the reaction between HX and the QZMNCPE, according to the method of Galus:39
| IC/IL = γ1/2[π1/2 erf (γ1/2) + exp (−γ)/γ1/2] | (3) |
| IC/IL = π1/2γ1/2 = π1/2 (kCbt)1/2 | (4) |
3.4. Calibration plot and limit of detection
Differential pulse voltammetry was used to determine the HX concentration (Fig. 5). Voltammograms clearly show that the plot of peak current versus HX concentration is comprised of two linear segments with different slopes (slope: 0.536 μA μM−1 for the first linear segment and 0.135 μA μM−1 for the second linear segment). These correspond to two different ranges of substrate concentration, including 1.0 to 100.0 μM for the first linear segment and 100.0 to 400.0 μM for the second linear segment. The decrease in sensitivity (slope) for the second linear range was likely due to kinetic limitation. The detection limit (3σ) for HX in the lower range region was found to be 0.173 μM. This value is comparable to those reported by other research groups. | ||
| Fig. 5 (A) Differential pulse voltammograms of QZTNMCPE in 0.1 M phosphate buffer solution (pH 7.0) containing different concentrations of HX. Curves a through f correspond to 1.0, 5.0, 10.0, 40.0, and 60.0 μM. (B) Plot of the electrocatalytic peak current as a function of HX concentration in the range of 1.0–100.0 μM and 100.0–400.0 μM obtained from differential pulse voltammograms. | ||
3.5 HX determination in real samples
To evaluate the applicability of the proposed method to real samples, it was applied to the determination of HX in water samples. The samples tested were found to be free from HX. Thus, synthetic samples were prepared by adding known amounts of HX to water samples. The results are given in Table 1.4. Conclusions
This work demonstrates the construction of a chemically modified carbon paste electrode by the incorporation of TiO2 nanoparticles and QZ as modifying species. The electrochemical behavior of QZ has been studied by cyclic voltammetry. This modified electrode shows very efficient electrocatalytic activity for the anodic oxidation of HX by substantially decreasing the anodic overpotential for this compound. A low detection limit, together with very easy preparation and regeneration of the electrode surface, long-term stability, and reproducibility, makes the system discussed above useful in the construction of simple devices for the determination of HX.The proposed electrode showed better selectivity than most previous electrodes and was proven suitable for the determination of HX in a real sample. This electrode is easy to prepare and use. It permits the direct measurement of HX in real samples without prior separation steps, considerably simplifying the determination procedure when compared to other applicable analytical methods.
Acknowledgements
The authors wish to thank the Yazd University Research Council, IUT Research Council, and Excellence in Sensors for financial support of this research. We gratefully acknowledge Dr N. Taghavinia of Sharif University of Technology for synthesising the nanoparticles.References
- T. Hoffman and H. Lees, The biochemistry of the nitrifying organisms, Biochem. J., 1953, 54, 579–583.
- R. P. Smith and W. R. Layne, A comparison of the lethal effects of nitrite and hydroxylamine in the mouse, J. Pharmacol. Exp. Ther, 1969, 165, 30–35 CAS.
- T. Kolasa and W. Wardencki, Quantitative determination of hydroxylamine, Talanta, 1974, 21, 845–857 CrossRef CAS.
- A. M. Prokai and R. K. Ravichandran, Simultaneous analysis of hydroxylamine, N-methylhydroxylamine and N, N-dimethylhydroxylamine by ion chromatography, J. Chromatogr., A, 1994, 667, 298–303 CrossRef CAS.
- F. Dias, A. S. Olojola and B. Jaselskis, Spectrophotometric determination of micro amounts of hydrazine and hydroxylamine alone and in the presence of each other, Talanta, 1979, 26, 47–49 CrossRef CAS.
- E. Kavlentis, Spectrophotometric determination of hydroxylamine using the DPGH-iron (II)-NH2OH ternary complex, Microchem. J., 1988, 37, 22–24 CrossRef CAS.
- A. Afkhami, T. Madrakian and A. Maleki, Indirect kinetic spectrophotometric determination of hydroxylamine based on its reaction with iodate, Anal. Sci., 2006, 22, 329–331 CrossRef CAS.
- X. Cui, L. Hong and X. Lin, Electrocatalytic oxidation of hydroxylamine on glassy carbon electrodes modified by hybrid copper–cobalt hexacyanoferrate films, Anal. Sci., 2002, 18, 543–547 CAS.
- M. Ebadi, Electrocatalytic oxidation of hydroxylamine by (RuPc)2 graphite modified electrode, Electrochim. Acta, 2003, 48, 4233–4238 CrossRef CAS.
- J. Zhang, Y.-H. Tse, W. J. Pietro and A. B. P. Lever, Electrocatalytic activity of N, N′, N′, N′-tetramethyl-tetra-3,4-pyridoporphyrazinocobalt(II) adsorbed on a graphite electrode towards the oxidation of hydrazine and hydroxylamine, J. Electroanal. Chem., 1996, 406, 203–211 CrossRef CAS.
- M. Mazloum-Ardakani and Z. Taleat, Investigation of electrochemistry behavior of hydroxylamine at glassy carbon electrode by indigocarmine, Int. J. Electrochem. Sci., 2009, 4, 694–706 Search PubMed.
- F. X. Dias and B. Jaselskis, Voltammetric determination of hydrazine and hydroxylamine, Analyst, 1983, 108, 76–80 RSC.
- P. E. Iversen and H. Lund, Polarographic determination of hydroxylamine and some hydrazine derivatives, Anal. Chem., 1969, 41, 1322–1324 CrossRef CAS.
- S. P. Mallela and B. L. Khandelwal, Use of the rotating platinum electrode for microdetermination of hydrazine, hydroxylamine, nitrite, ascorbic acid, oxalic acid and thiourea with manganese(III) pyrophosphate, Microchim. Acta, 1977, 68, 245–248 CrossRef.
- M. Yang and J. J. Zhu, Indirect voltammetric determination of trace hydroxylamine using magnetic microspheres, Analyst, 2003, 128, 178–181 RSC.
- M. Mazloum Ardakani, M. A. Karimi, S. M. Mirdehghan, M. M. Zare and R. Mazidi, Electrocatalytic determination of hydroxylamine with alizarin red S as a homogenous mediator on the glassy carbon electrode, Sens. Actuators, B, 2008, 132, 52–59 CrossRef.
- J. Li and X. Lin, Electrocatalytic oxidation of hydrazine and hydroxylamine at gold nanoparticle-polypyrrole nanowire modified glassy carbon electrode, Sens. Actuators, B, 2007, 126, 527–535 CrossRef.
- M. Uddin Ahmed, M. Mosharraf Hossain and E. Tamiyaa, Electrochemical biosensors for medical and food applications, Electroanalysis, 2008, 20, 616–626 CrossRef.
- R. De Marco, G. Clarke and B. Pejcicb, Ion-selective electrode potentiometry in environmental analysis, Electroanalysis, 2007, 19, 1987–2001 CrossRef CAS.
- M. H. P. Santana, L. M. D. Silva, A. C. Freitas, J. F. C. Boodts, K. C. Fernandes and L. A. De Faria, Application of electrochemically generated ozone to the discoloration and degradation of solutions containing the dye reactive orange 122, J. Hazard. Mater., 2009, 164, 10–17 CrossRef CAS.
- K. Pihel, T. J. Schroeder and R. Mark Wightman, Rapid and selective cyclic voltammetric measurements of epinephrine and norepinephrine as a method to measure secretion from single bovine adrenal medullary cells, Anal. Chem., 1994, 66, 4532–4537 CrossRef CAS.
- S. Shahrokhian and M. Amiri, Multi-walled carbon nanotube paste electrode for selective voltammetric detection of isoniazid, Microchim. Acta, 2007, 157, 149–158 CrossRef CAS.
- H. Beitollahi, M. Mazloum Ardakania, B. Ganjipour and H. Naeimi, Novel 2,2′-[1,2-ethanediylbis(nitriloethylidyne)]-bis-hydroquinone double-wall carbon nanotube paste electrode for simultaneous determination of epinephrine, uric acid and folic acid, Biosens. Bioelectron., 2008, 24, 362–368 CrossRef CAS.
- M. Mazloum-Ardakani, H. Beitollahi, B. Ganjipour, H. Naeimi and M. Nejati, Electrochemical and catalytic investigations of dopamine and uric acid by modified carbon nanotube paste electrode, Bioelectrochemistry, 2009, 75, 1–8 CrossRef CAS.
- M. A. El Mhammedi, M. Achak, M. Hbid, M. Bakasse, T. Hbid and A. Chtaini, Electrochemical determination of cadmium(II) at platinum electrode modified with kaolin by square wave voltammetry, J. Hazard. Mater., 2009, 170, 590–594 CrossRef CAS.
- W. Sun, X. Li, Y. Wang, X. Li, C. Zhao and K. Jiao, Electrochemistry of myoglobin in Nafion and multi-walled carbon nanotubes modified carbon ionic liquid electrode, Bioelectrochemistry, 2009, 75, 170–175 CrossRef CAS.
- G. P. Jin, Q. Z. Chen, Y. F. Ding and J. B. He, Electrochemistry behavior of adrenalin, serotonin and ascorbic acid at novel poly rutin modified paraffin-impregnated graphite electrode, Electrochim. Acta, 2007, 52, 2535–2541 CrossRef CAS.
- M. A. El Mhammedi, M. Bakasse and A. Chtaini, Electrochemical studies and square wave voltammetry of paraquat at natural phosphate modified carbon paste electrode, J. Hazard. Mater., 2007, 145, 1–7 CrossRef CAS.
- Y. X. Sun, S. F. Wang, X. H. Zhang and Y. F. Huang, Simultaneous determination of epinephrine and ascorbic acid at the electrochemical sensor of triazole SAM modified gold electrode, Sens. Actuators, B, 2006, 113, 156–161 CrossRef.
- O. Lev, Z. Wu, S. Bharathi, V. Glezer, A. Modestov, J. Gun, L. Rabinovich and S. Sampath, Sol–gel materials in electrochemistry, Chem. Mater., 1997, 9, 2354–2375 CrossRef CAS.
- M. Mazloum Ardakani, P. Ebrahimi karami, P. Rahimi, H. R. Zare and H. Naeimi, Electrocatalytic hydrazine oxidation on quinizarine modified glassy carbon electrode, Electrochim. Acta, 2007, 52, 6118–6124 CrossRef.
- G. Y. Kim, J. Shim, M. S. Kang and S. H. Moon, Optimized coverage of gold nanoparticles at tyrosinase electrode for measurement of a pesticide in various water samples, J. Hazard. Mater., 2008, 156, 141–147 CrossRef CAS.
- M. Chikae, T. Fukuda, K. Kerman, K. Idegami, Y. Miura and E. Tamiya, Amyloid-β detection with saccharide immobilized gold nanoparticle on carbon electrode, Bioelectrochemistry, 2008, 74, 118–123 CrossRef CAS.
- J. H. Chang, T. J. Yang and C. H. Tung, Performance of nano- and nonnano-catalytic electrodes for decontaminating municipal wastewater, J. Hazard. Mater., 2009, 163, 152–157 CrossRef CAS.
- M. Mazloum Ardakani, Z. Taleat, H. Beitollahi, M. Salavati-Niasari, B. B. F. Mirjalili and N. Taghavinia, Electrocatalytic oxidation and nanomolar determination of guanine at the surface of a molybdenum (VI) complex–TiO2 nanoparticle modified carbon paste electrode, J. Electroanal. Chem., 2008, 624, 73–78 CrossRef CAS; Z. Hosseini, N. Taghavinia, N. Sharifi, M. Chavoshi and M. Rahman, Fabrication of high conductivity TiO2/Ag fibrous electrode by electrophoretic deposition method, J. Phys. Chem. C, 2008, 112, 18686–18689 CAS.
- E. Laviron, General expression of the linear potential sweep voltammogram in the case of diffusionless electrochemical systems, J. Electroanal. Chem., 1979, 101, 19–28 CrossRef CAS.
- A. J. Bard, L. R. Faulkner, Electrochemical Methods, Fundamentals and Applications, Wiley, New York, 2001 Search PubMed.
- C. P. Andrieux and J. M. Saveant, Heterogeneous (chemically modified electrodes, polymer electrodes) vs. homogeneous catalysis of electrochemical reactions, J. Electroanal. Chem., 1978, 93, 163–168 CrossRef CAS.
- Z. Galus, Fundamentals of Electrochemical Analysis, Ellis Horwood, New York, 1976 Search PubMed.
| This journal is © The Royal Society of Chemistry 2010 |