NIR diffuse reflectance for on-scale monitoring of the polymorphic form transformation of pazopanib hydrochloride (GW786034); model development and method transfer
Susan E.
Barnes
*a,
Thomas
Thurston
b,
Jo Ann
Coleman
c,
Ann
Diederich
a,
Darryl
Ertl
a,
James
Rydzak
d,
Patrick
Ng
e,
Katherine
Bakeev
f and
Dharmesh
Bhanushali
d
aChemical Development, GlaxoSmithKline, 5 Moore Drive, Research Triangle Park, NC 27709, USA. E-mail: Susan.E.Barnes@gsk.com
bChemical Development, GlaxoSmithKline, Old Powder Mills, Leigh, Tonbridge, Kent TN11 9AN, UK
cStatistical Sciences, GlaxoSmithKline, 2301 Renaissance Blvd, King of Prussia, PA 19406, USA
dChemical Development, GlaxoSmithKline, 709 Swedeland Road, King of Prussia, PA 19406, USA
eGlobal manufacturing and Supply, GlaxoSmithKline, Jurong, Singapore
fCAMO Software Inc., One Woodbridge Center, Suite 319, Woodbridge, NJ 07095, USA
First published on 30th September 2010
Abstract
We report the successful application of in situ diffuse reflectance near-infrared (NIR) spectroscopy for characterization of the solvent mediated polymorphic form transformation of pazopanib hydrochloride. Crystal form is a critical quality attribute (CQA) for this active pharmaceutical ingredient (API) to ensure appropriate levels of exposure in patients. NIR spectroscopy was implemented to monitor this final stage unit operation in real-time on manufacturing scale. This technique provided an automated in situ method to replace labour intensive off-line qualitative testing and manual sampling. A partial least squares (PLS) model was created using NIR data collected during an early manufacturing campaign and a novel strategy based on interval-PLS was used to select and optimize the model parameters. The model was subsequently implemented on plant scale and used to monitor clinical manufacturing campaigns. The PLS method clearly differentiated between the monohydrate and the desired anhydrous form allowing determination of the transformation end point in real-time. Utilisation of PLS diagnostics such as Mahalanobis distance and spectral residuals provided additional capability to signal any unexpected behaviour in the process and/or indicate the presence of an undesired solvate polymorph also seen during process development.
1. Introduction
The aim of chemical development is to design robust, cost effective manufacturing processes that consistently deliver superior active pharmaceutical ingredients (APIs) with pre-defined quality and performance. Quality cannot be tested into products it must be built-in or should be by design.1 Knowledge gained from research and development studies in conjunction with manufacturing experience provide scientific understanding for determination of the design space and manufacturing controls required to meet the desired product specifications.2 Process Analytical Technologies (PAT) are systems for designing, analyzing, and controlling pharmaceutical manufacturing processes. Implementation of PAT allows timely measurement of critical quality and performance attributes of raw and process materials. Incorporation of PAT into quality by design (QbD) encompasses the use of scientifically based process optimization, sensor technologies, statistical tools (chemometrics), knowledge management and feedback process control to ensure manufacture of high quality products.1,2The manufacture of quality drug substance frequently involves isolation and recovery of the solid API where crystallization plays a key role as a separation and purification unit operation.3,4 The final crystallization process should provide firm control of particle size distribution, particle morphology and polymorphic form, all of which can be critical quality attributes (CQAs) of a drug substance.5 Frequently a chemical entity will have multiple crystal polymorphs that can exhibit different physical and biological properties, hence production of the correct polymorph is therefore essential.6 Taking a QbD approach to creating the final particle forming step is central to form control and critical to ensuring product stability, safety, efficacy and performance at secondary manufacturing.7
Traditionally off-line techniques such as X-ray powder diffraction (XRPD), differential scanning calorimetry (DSC), solid state NMR and infrared spectroscopy (IR) are used in polymorph testing.6–8 However, these off-line tests are not available in real-time, can be labor intensive and may potentially give misleading results in cases where the sample is unstable when removed from process conditions. Additionally, off-line testing only provides single time-point measurements that do not fully profile the transformation kinetics. Application of PAT can provide a greater understanding of the thermodynamics and kinetics of a crystallization process, and could potentially lead to precision manufacture of the desired crystal form.9–11 There is a continuing drive toward implementation of PAT for crystallization and form monitoring on larger scale to assess scale-up effects (such as change in cooling rate, heat transfer effects, reactor and baffle geometry) that may impact the properties of the final API.12,13 The application of PAT on manufacturing scale offers many other potential benefits including reduced cycle times, increased safety for plant operators, reduction in material waste and reduction in time/labor for off-line laboratory analysis.
A number of in situ technologies have been employed on-line at laboratory scale as tools for monitoring crystallizations and crystal form. In situ mid-infrared (MIR) spectroscopy is frequently used for measurement of supersaturation during solution crystallization processes.14–16 The supersaturation is the driving force of any cooled crystallization process and thus a key parameter to monitor for control of end product properties. Focused beam reflectance measurements (FBRM) have been reported to provide information on chord length distribution during crystallization processes.17,18 FBRM can also be applied during form conversion processes if the polymorphic transition is accompanied by a distinct change in crystal habit.19 Raman spectroscopy is widely used as an in situ technique for analysis of crystalline form20–24 and is especially effective for analysis of solvent mediated transformations due to its low sensitivity to aqueous solvents.25 Both qualitative and quantitative models have been applied to Raman data in order to map and often quantify the solute concentration as a function of the conversion time.23 The application of fiber-optic near-infrared (NIR) spectroscopy for monitoring crystal form in a slurry has also been reported in the literature.26–30 The application of multivariate modeling to NIR process data has increased and broadened the application of this technique in monitoring API chemistries and pharmaceutical production processes.31–34 The robustness of calibration transfer,30,35–39 use of fiber optic technology and rapid measurement rates makes NIR an attractive in-process monitoring tool. Thus, NIR spectroscopy is at the forefront of efforts to implement PAT for continuous quality assurance/real-time release.1,2
The work presented herein details the application of in situ FT-NIR diffuse reflectance spectroscopy to monitor the final stage solvent mediated form transformation of pazopanib hydrochloride on manufacturing scale.40 Pazopanib hydrochloride (Fig. 1) is a potent and selective inhibitor of human VEGFR2 tyrosine kinase and is being developed as an oral agent for oncology indications such as advanced or metastatic renal cell carcinoma (RCC) and other solid tumors.41 Following laboratory and pilot level based proof of concept work, an NIR diffuse reflectance probe was installed at manufacturing scale for monitoring of the monohydrate to anhydrate form transformation. Three forms of the API were observed during process development: the intermediate monohydrate form, the final anhydrous API and an undesired acetonitrile (ACN) solvate.40 NIR was shown to be an effective in situ technology for identification of all three crystalline forms in solution and allowed for real-time monitoring of the form transformation. NIR data were collected on manufacturing scale for information only to gather more process understanding. Transfer of the NIR technique into manufacturing required focus on two aspects: (1) hardware installation and design/commissioning of interfaces that are compatible and non-invasive into the process, and (2) creation, transfer and validation of methods used for real-time data trending. In situ NIR data and off-line XRPD data were collected for two manufacturing campaigns. Partial least squares (PLS)41,43 and a novel model optimization approach were implemented for real-time analysis and diagnosis of the data from the form conversion process.
 | ||
| Fig. 1 Pazopanib hydrochloride form transformation and crystallization process.40 | ||
2. Experimental
2.1 Manufacturing scale form transformation: process description
Polymorph screening studies of the pazopanib hydrochloride drug substance identified solvates formation with numerous solvent classes. As a result of the screening process the final particle forming step of the current API synthesis was developed in acetonitrile (ACN)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water.40 The API forming stage was developed in two parts utilizing the monohydrate form of the API as an intermediate to gain control over drug substance attributes including product purity, appearance and polymorphic form (Fig. 1). The first part of the API forming stage was a re-crystallization of the anhydrate (1) from aqueous acetonitrile to generate the monohydrate form (1a) of pazopanib hydrochloride. The second part was the dehydration of the monohydrate to the desired anhydrous polymorph (1).40 Dehydration to the anhydrate was processed in an agitated filter where hot seeded aqueous acetonitrile was charged to the monohydrate in the dryer at >70 °C. The resulting slurry was stirred continuously until the form transformation to the anhydrate was complete.
:water.40 The API forming stage was developed in two parts utilizing the monohydrate form of the API as an intermediate to gain control over drug substance attributes including product purity, appearance and polymorphic form (Fig. 1). The first part of the API forming stage was a re-crystallization of the anhydrate (1) from aqueous acetonitrile to generate the monohydrate form (1a) of pazopanib hydrochloride. The second part was the dehydration of the monohydrate to the desired anhydrous polymorph (1).40 Dehydration to the anhydrate was processed in an agitated filter where hot seeded aqueous acetonitrile was charged to the monohydrate in the dryer at >70 °C. The resulting slurry was stirred continuously until the form transformation to the anhydrate was complete.
2.2 Laboratory scale experimentation
A number of proof-of-concept laboratory experiments were conducted on 1 L scale using a Mettler Toledo automatically controlled LabMax reactor system. Form transformation experiments were conducted following the process detailed in section 2.1. Particle morphology was investigated using optical microscopy under cross-polar filters (Olympus BX51). FT-NIR spectra were collected using a Bruker Matrix-F spectrometer (Bruker Optics) qualified for use with a Hastelloy ¼ “diameter 16” long Hastelloy diffuse reflectance probe with a sapphire window and 3 m of fiber optics.2.3 In situ NIR diffuse reflectance probe set-up: manufacturing scale
For acquisition of in situ form conversion data at manufacturing scale, a NIR diffuse reflectance probe was custom-designed to interface with the processing equipment in the manufacturing plant (Fig. 2). The Hastelloy optical probe was designed with nitrogen purge capability with purge ports positioned such that nitrogen could be driven across the front of the sapphire window to prevent probe fouling in situ. The probe was inserted into a custom-built flange allowing installation into a sampling port on the manufacturing equipment. The probe was seated flush with the wall of the filter dryer so as not to hinder the path of the agitator blades during processing. | ||
| Fig. 2 NIR diffuse reflectance probe (A) with nitrogen purge ports inserted into the filter dryer interface (B) before connection to the filter dryer. | ||
FT-NIR spectra were collected using a Bruker Matrix-F spectrometer (Bruker Optics) connected to the probe via 50 m of steel reinforced optical fiber. Installation qualification (IQ), operational qualification (OQ) and performance qualification (PQ) of the NIR instrument and diffuse reflectance probe were conducted at the manufacturing site.
NIR data were collected on scale over the course of two manufacturing campaigns, each campaign being greater than 10 batches. Data were collected for all campaign batches and are presented herein. NIR data were used for information only and were collected in conjunction with off-line XRPD data to confirm agreement between the on-line and off-line measurements.
2.4 NIR data acquisition and data analysis
NIR data were collected and analyzed using Bruker OPUS software Version 6.5 in the wavelength region 12000–4000 cm−1 at 8 cm−1 resolution averaging 32 scans. On manufacturing scale the OPUS software was interfaced to the plant distributed control system (DCS) and to Aspentech IP21 Process Historian Database for real-time, remote viewing of the PLS model results alongside the physical process data (Fig. 3). Model development studies were conducted using Matlab R2006a (The Mathworks, Natick, Massachusetts), and an in-house software package was used for real-time Principal Components Analysis.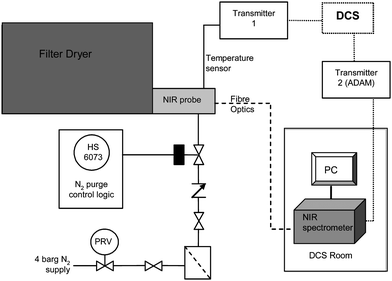 | ||
| Fig. 3 Schematic of NIR set-up in Global Manufacturing and Supply. | ||
During the first campaign qualitative process profiles were obtained in real-time by applying Principal Components Analysis (PCA)42 to each individual batch of data. PCA was selected because it has the advantage of not requiring any calibration reference spectra which, prior to this campaign, were not available from the plant instrumentation. Prior to applying PCA, data were pre-treated by calculating second-derivative spectra (using a 17-point Savitsky–Golay filter)43 followed by mean-centering and selection of two spectral regions 6250–6000 cm−1 and 5050–5150 cm−1. Lab studies had previously shown these regions to encompass bands characteristic of the monohydrate and solvate forms.
Spectra from the first campaign were used as reference data to create a semi-quantitative calibration model for application in later campaigns. A key consideration during model development was the limited nature of reference data available from the process. Mid-process off-line reference samples could not be taken because of the potential of producing the incorrect form under the rapid cooling conditions created by removing the sample from the vessel prematurely. Rapid conversion also prevented the use of prepared calibration standard mixtures. No representative plant-scale reference spectra of the ACN solvate polymorph were available during model building. Consequently, the only spectra with known composition at plant-scale were those of 100% monohydrate (before the form conversion started), and those of 100% anhydrate (after the form conversion was complete). These spectra were considered suitable as calibration standards of 100% monohydrate and 100% anhydrate respectively. For modelling purposes these were assigned “% conversion” values of 0% and 100% respectively.
Partial least squares (PLS)42 was selected for analysis of data generated in subsequent campaigns. Model parameters, specifically data pre-treatment and wavelength range, were selected using a novel Matlab based algorithm based on interval-PLS.44 In the variation used in this work, the NIR spectrum is split into several adjacent intervals with boundaries sited based on features in the spectra (Fig. 4).
 | ||
| Fig. 4 Spectral intervals with first-derivative monohydrate and anhydrate reference spectra overlaid. | ||
An exhaustive search was then conducted over all possible combinations of the wavelength intervals with the objective of finding which combinations best met some pre-defined criteria. This is repeated for different types of data pre-treatment. In this case the NIR spectrum was split into 13 regions, and 5 different types of pre-treatment were considered, meaning that 5 × (213 − 1) = 40955 different sets of model parameters were compared using this approach.
The approach used to evaluate performance of each combination of model parameters is outlined in Fig. 5. For each possible scenario (combination of pre-treatment type and wavelength regions), reference spectra from 4 batches were used as a calibration set and the model rank evaluated by an internal leave-one-batch-out cross-validation (LOBO-CV). A temporary model is then built using the rank that gave the lowest LOBO-CV error. An additional 3 batches are used as an external test set to obtain an overall Root Mean Square Error of Prediction (RMSEP) for this model, plus individual RMSEPs for the monohydrate and anhydrous forms separately.
 | ||
| Fig. 5 Schematic of the screening approach used to determine the optimum pre-processing and wavelength combination for the final PLS model. | ||
Fig. 6 plots the RMSEPs of the highest-performing scenarios, coloured by pre-treatment type; points towards the lower-left corner of this plot correspond to model parameters that have the lowest prediction error.
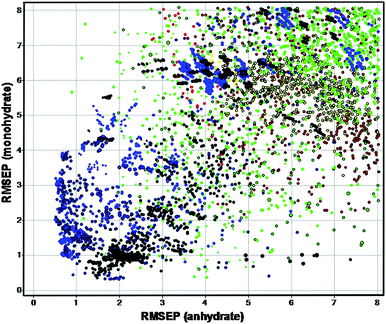 | ||
| Fig. 6 Graphical comparison of model performance for different scenarios (colour represents pre-processing type); promising scenarios are then selected for further evaluation. | ||
Several of the top-performing scenarios were then selected for further comparison by the process expert, thus bringing human expertise back into the model building process. A combination of first-derivative + standard normal variate (SNV)45 pre-processing with wavelength intervals 1–4, 8 and 11 was selected. Additionally, in order to increase the method's ability to detect the ACN solvate, should it be formed in the process, a portion of interval 5 was included in the final model. Comparison of the RMSEP before and after this adjustment confirmed that sensitivity to the monohydrate and anhydrate forms had not been significantly affected. The model contained 4 PLS factors.
The final model was constructed using data from all batches from campaign 1, of which four were set aside as an external test-set. Results from the external test set batches were used to establish a statistically derived prediction limits for the model-predicted % conversion value that can be considered sufficient to indicate that the form transformation process is complete. The lower and upper 95% prediction limits for the predicted conversion % were 99.12 and 100.46% respectively. Upper limits were also established for the Mahalanobis distance (<0.048) and spectral-residuals diagnostics (<0.059).
These values act as diagnostic tools to assess the similarity between the current NIR spectrum of the process, and the reference spectra used to create the PLS model. These limits for individual spectra are intended to be used to assess batch completion by detecting the first spectra that comply with all three limits.
2.5 Assessment of limit-of-detection and limit-of-quantification
Limit of detection (LOD) and limit of quantification (LOQ) of the NIR application were estimated using a partial least squares (PLS) model of spectral data. Data were obtained from a series of laboratory based dosing experiments which ran under modified conditions to ensure stability of each form being monitored. The experiments were designed to ensure that the NIR probe was exposed to samples that were representative of the slurry seen on manufacturing scale. To assess the sensitivity of the NIR to the presence of residual monohydrate in the slurry, small amounts of the monohydrate were gravimetrically dosed into the slurry of the anhydrate at room temperature. NIR spectra were acquired at each concentration and a PLS model of the spectra and associated monohydrate concentrations (0.0–4.5% w/w) were created to estimate the LOD and limit of quantification (LOQ) for the NIR. Data were collected in OPUS and the PLS models were built using OPUS Quant software. The same spectral regions and pre-processing type as those used in the main form transformation model were applied. For the purpose of this study the LOD and LOQ were defined as being 3× RMSEP and 10× RMSEP respectively.3. Results
3.1 In situ NIR data from the pazopanib hydrochloride final stage form conversion
Three forms of pazopanib hydrochloride were observed to be stable in an acetonitrile/water solvent system during laboratory scale development work. These were the monohydrate (used as an intermediate in the final stage), the desired anhydrate and an undesired acetonitrile (ACN) solvate.40 Each crystal form has a very distinctive crystal habit, powder X-ray diffraction pattern and off-line FTIR and Raman spectrum. Optical microscope images of the three forms of the API are shown in Fig. 7. The morphology of the crystalline anhydrate is equant whereas that of the monohydrate is acicular and the ACN solvate is lath. | ||
| Fig. 7 Optical microscope images of (A) monohydrate, (B) anhydrate and (C) ACN solvate forms of pazopanib hydrochloride. | ||
Fig. 8 is an overlay of NIR spectra of each crystal form individually slurried in 6 volumes 97:3 ACN:water. The spectrum of the monohydrate contains a prominent shoulder at 5100 cm−1/1958 nm ascribed to the bound water in the crystal lattice (STP 4% water).46 Although features from the free water in the solvent system are observed in the spectrum (5250 cm−1/1906 nm) they do not mask the bound water band of the monohydrate. Distinct differences in the spectra of all three forms are present in the CH 1st overtone region (6250–6000 cm−1 and 1600–1800 nm) which are not masked by solvent features. The monohydrate, anhydrate and ACN solvate have bands at 6116 cm−1, 6095 cm−1 and 6080 cm−1 respectively which can be used for visual identification of each form in situ.
 | ||
| Fig. 8 Overlay of NIR spectra in slurry; monohydrate, anhydrate and ACN solvate. | ||
Based on knowledge gained from the NIR data acquired in the laboratory, a PCA method was used for trending the form conversion at manufacturing scale. Following technology transfer of the PCA method from R&D to manufacturing, conversion data were collected and trended throughout a clinical manufacturing campaign. Fig. 9 is an overlay of the raw data as a function of time for a form conversion batch on manufacturing scale.
 | ||
| Fig. 9 3D profile of form transformation from hydrate to anhydrous form. | ||
Spectra from the manufacturing process were of excellent quality and signal to noise. A gradual reduction in the intensity and eventual disappearance of the band at 5100 cm−1 can be observed as the batch converts from monohydrate to anhydrate. Throughout the conversion a shift in the C–H 1st overtone band at 6116 cm−1 to 6095 cm−1 is seen. From the raw spectral data, it is clear the conversion rapidly occurs directly from the monohydrate to the anhydrate. As expected there is no indication that the ACN solvate forms at any point during the process.
Fig. 10 is an overlay of a scores plot for the first principal component of the NIR PCA method extracted for an individual campaign batch. The kinetic profile of the conversion as a function of time is overlaid with the cake temperature. The scores clearly trend upward as the conversion progresses and show that the transformation is complete after 75 minutes. The temperature profile indicates that the vessel temperature was maintained above 70 °C as written in the manufacturing process working directions.40 In this instance, the vessel temperature was lowered to 65 °C after 500 minutes to extract a sample for off-line XRPD analysis (for-information-only). The XRPD sample was in agreement with the NIR data that the form transformation was complete. For all campaign batches the NIR data and the PCA scores plot showed agreement with for-information-only XRPD off-line samples. The PCA scores for all batches clearly confirmed completion of the transformation, however, the scores alone did not give molecular information on the form present.
 | ||
| Fig. 10 NIR PCA scores plot (PC1) and slurry temperature data acquired during a manufacturing scale form transformation. | ||
3.2 Limits of detection and quantification
The LOD and LOQ for the monohydrate in the binary mixture were 0.44% w/w and 1.47% w/w respectively. A similar dosing experiment was carried out for determination of the sensitivity of the NIR to the presence of ACN solvate in a slurry of the anhydrous form (0.0–9.5% w/w ACN solvate). The calculated LOD and LOQ for the solvate were 0.41 wt% and 1.36 wt% respectively. Data were collected for both models using the laboratory based NIR and probe and thus the LOD values calculated could not be directly applied to the spectral data acquired on plant scale. However, the calculated values served as a guide for maximum possible instrument sensitivity in accordance with the process.3.3 Model application
The optimised PLS model (Section 2.4) was used for analysis during a second manufacturing campaign. Connection of the OPUS software to the DCS and IP21 allowed real-time profiling of the PLS model predictions and Mahalanobis distance alongside the physical data from the vessel. The model transferred well to this campaign. Fig. 11 is a plot of the model predictions, Mahalanobis distance and X-residual values for a single batch. | ||
| Fig. 11 Model predictions and diagnostics for one batch from manufacturing campaign. | ||
Fig. 11 illustrates that for this particular batch, the predicted % conversion values finish close to the limits and the model diagnostics fall below or close to the established limits. Table 1 shows a summary of the compliance of the NIR data from each batch with the set limits of the model. Some deviation from the limits was observed over the batches, which was ascribed to the limited quantity of data that was available to build the model (all of which came from a single campaign). The values presented are the first compliant individual spectra from 20 spectra recorded after off-line XRPD test method confirmed complete conversion. Highlighted data from four (out of 10) batches did not meet at least one of the three statistical limits (three for the predicted % conversion, and one for spectral-residuals), although the discrepancy is small as seen in Table 1 were the values of the closest spectra as compared to the limits are also shown. The differences indicate that the model should be updated to include further campaign data which is anticipated to allow it to better cope with campaign-to-campaign variability in the future.
| Batch # | Predicted % conversion | M-Distance | Spectral residuals |
|---|---|---|---|
| 1 | 100.7 | 0.003 | 0.0401 |
| 2 | 100.45 | 0.0051 | 0.0314 |
| 3 | 100.81 | 0.0029 | 0.0389 |
| 4 | 100.34 | 0.013 | 0.0539 |
| 5 | 100.3 | 0.03 | 0.0456 |
| 6 | 100.32 | 0.0025 | 0.0709 |
| 7 | 100.21 | 0.0051 | 0.0411 |
| 8 | 100.31 | 0.0078 | 0.0318 |
| 9 | 100.15 | 0.0098 | 0.0414 |
| 10 | 100.49 | 0.011 | 0.0582 |
| Limits | 99.12 to 100.46 | 0.048 | 0.059 |
The same PLS model was applied during another campaign ran on smaller pilot plant (PP) scale using the same working directions. Here the NIR data were collected from a smaller scale filter using a different Bruker-Matrix FT-NIR spectrometer and a different NIR diffuse reflectance probe. In common with the manufacturing-scale installation, the probe was located flush with the internal wall of the vessel to avoid contact with the agitator blades. Fig. 12 shows conversion profiles of several batches from the second manufacturing campaign in comparison to data from the smaller-scale pilot plant campaign.
 | ||
| Fig. 12 Comparison of PLS transformation profiles from two manufacturing batches and two pilot plant batches. | ||
The model transferred well to application in the PP campaign without additional spectra being incorporated into the model. For both campaigns the data indicated that the form conversion process reached completion several hours before the off-line sample would normally be taken thus indicating a cycle time reduction may be possible.
4. Discussion
4.1 NIR form transformation data
In situ NIR spectroscopy proved to be a successful technique for real-time trending of the monohydrate to anhydrate form conversion. For this application each of the crystal forms of interest had distinct, separately identifiable features in the NIR data which were not masked by the solvent bands. This may not be the case in all systems where individual bands for each form are not present or not seen due to presence of broad solvent bands.Good quality spectra of high signal-to-noise were acquired from the diffuse reflectance probe (and 50 m fiber optics) indicating good contact of the raw material with the probe. Raw spectral data had clear features for identification of both expected polymorphic forms. For each of the campaign batches, agreement was seen between the off-line for-information-only XRPD data and the NIR in situ data, each confirming completion of the conversion process to the anhydrate. The data demonstrate that the current process is robust, consistently producing the desired crystal form. Although data are currently collected for process information only, the results indicate that there may be a future opportunity to implement the NIR to monitor the conversation process leading to a possible reduction in the cycle time of the conversion step, thus allowing cost and energy savings. This may also lead to labor savings of several hours associated with the time taken for off-line sampling and analysis. A further benefit to the model was the use of model diagnostics that provide the opportunity to detect any process deviations and confirm the absence of unexpected material during processing.
4.2 Model development
The application of PCA for data trending in the first manufacturing campaign was successful, however, there were a number of limitations from using a solely qualitative technique. To choose a more suitable methodology for NIR data analysis the particular challenges of this PAT application were considered.Since reliable reference spectra were only available at 0% and 100% conversion, two modelling approaches were considered for the data: a Conformity Index and a two-level PLS calibration model. The Conformity Index (CI)33,47,48 is obtained by first calculating the difference between the current spectrum and the average of some reference spectra. This is then scaled by dividing by the standard deviation of the reference spectra on a wavelength-by-wavelength basis, and taking the largest absolute value of this result as the CI. This approach is well suited for industrial raw material and intermediate qualification and identification as it removes the need for exhaustive calibration work.32 However, in this application, it allows the use of only the anhydrate form as the reference material in a single model.
Due to these limitations, PLS was selected as the technique to use for modelling further campaign data.42,44 PLS calibrations are usually built with reference standards at several different concentration values, whereas in this application standards were only available at two levels (0% and 100% conversion). As a result, it is reasonable to expect that this model will give accurate results only when the form conversion is just starting or close to completion; predicted % conversion values mid-way through the form conversion are likely to be indicative of progress but are not likely to be accurate. However, this limitation is not restrictive for this application, as accuracy is only critical as the process reaches completion. Utilisation of the Mahalanobis distance and spectral-residuals PLS diagnostics also provided additional capability to signal any unexpected behaviour in the process and/or indicate the presence of the undesired solvate form.30,31 Mahalanobis distance itself is commonly used as a technique to qualify and provide discrimination between samples differing in physical and/or chemical properties as well as its use in materials characterisation for quality control purposes.49,50
For validation purposes, it was important to assess the accuracy of the final PLS model for detection of acceptable and unacceptable samples.47 Four batches were set aside as an external test set in order to validate the model for analysis of data from other acceptable batches. Results from this test set were used to establish the statistical tolerance intervals for the model. A further test of model robustness was to determine its sensitivity to NIR spectra of unexpected material. Although the process conditions were developed according to a Quality by Design philosophy, thus mitigating the risk of undesired acetonitrile solvate formation, the solvate was the only available material of any relevance which could be used in such a test. Hence the model was tested against solvate spectra from the laboratory. The results indicated that the PLS % conversion trend alone may not be sufficient to detect unexpected material since the presence of the solvate only resulted in a reduction in the predicted % conversion, which is also seen when monohydrate material is present. However, the model diagnostics increased significantly with the presence of the ACN solvate in the slurry spectra. Both the M-distance (0.6) and spectral residuals (0.45) fell appreciably outside of the set statistical limits (0.048 and 0.059 respectively) demonstrating that unexpected material can be easily detected by the use of these diagnostics.
4.3 Model update
The construction of multivariate calibrations is often laborious due to the requirement to base the model on a sufficiently broad group of representative samples. The number of samples available, overall characteristics of the samples to include, wavelength and pre-treatment selection and choice of the most representative data, are all factors that can make the calibration process complex.31 Here we presented a novel optimisation strategy for the choice of model pre-treatment and wavelength selection which was less labour intensive than other more traditional approaches. In addition, the use of a two-level PLS modeling as opposed to fully quantitative analysis removed the need for time consuming calibration construction using a larger sample set.It makes sense to consider chemometric models that can be updated so that they can continue to be used after modifications to the production cycle or to the instrument.51,52 A way of accounting for new variations in the spectral data from the process is to rebuild the model with addition of further samples to the original calibration set.35 The addition of suitable new samples can make the model more robust to natural variability in measurement and process conditions, and often leads to more robust predictions. Model updating can generally be applied when instrumental and process differences are not too complicated or extreme. In general, models should be checked regularly and attention given to the model diagnostics (M-distance and spectral-residuals) in order to map model and instrument performance over time.34 Any new variability sources that can cause significant calibration errors should be included in the model.30 These include differences in the hardware and differences in spectrometer response (spectral bandwidth, wavelength calibration, etc.). Of course many of these factors can also be mitigated by routine instrument maintenance and upkeep.
Application of the model to the second manufacturing campaign dataset showed slight differences in the predicted % conversion values, Mahalanobis distance and spectral-residuals over the 10 batches. Some deviation from the pre-defined tolerance intervals and diagnostic limits was expected, due to the limited quantity of data available to build the model (all of which came from a single campaign). On interrogation of the raw data no obvious chemical or physical process changes were observed and thus the differences between model diagnostics for the two campaigns were ascribed to slight changes in spectrometer/probe/fiber performance, and/or removal and re-installation of the probe. The differences indicate that the model should be updated to include data from the more recent manufacturing campaign and the statistical limits re-calculated. In the long term, it is expected that further updating and maintenance of the model with new data from additional campaigns will add to the model's accuracy and robustness.
4.4 Model transfer
Since construction of a calibration model can be time intensive, it is often desirable to be able to transfer a model from one instrument to another. There are an extensive number of reports in the literature on NIR calibration transfers documenting different approaches to solving calibration transfer issues.30,35–37 A range of studies have concluded that qualitative models are easier to transfer than quantitative models. Whilst multivariate models built on broad wavelength regions of the NIR spectrum have been demonstrated to offer the ability to handle complex mixtures with better sensitivity, these models typically exhibit a greater dependence on the instrument's measurement reproducibility compared to qualitative methods or models that employ data over selected wavelengths.30In the current study, the model built from manufacturing scale data was transferred to a similar but independent system in the pilot plant and in both cases the PAT application was a suitable indicator of process completion. The model transferred well and produced comparable results irrespective of scale, shown by the similarity in predictions and model diagnostics from both manufacturing and pilot plant scale. In this case, no further update of the model was required showing good transferability of the model between NIR equipment and scale. However, for long term use a separate model specific to the pilot plant installation may be developed. These data are compatible with general expectations for on-line measurements where contact between the probe and the process content is generally good and scale-independent.
5. Conclusions
In situ NIR diffuse reflectance spectroscopy has been shown to be a successful technique for monitoring of the monohydrate to anhydrate form conversion of pazopanib hydrochloride (GW786034B) on manufacturing scale.A two-level PLS calibration was developed to trend the form transformation in real-time during processing. Use of the calibration was shown to be a significant improvement over PCA analysis of the data. An exhaustive search approach based on interval-PLS was used to screen a wide range of model parameters in order to develop a robust model for process monitoring. This novel technique is a more cost effective and less labor intensive approach to wavelength and region selection in comparison to traditional manual optimization.
Implementation of NIR spectroscopy and PLS model to monitor the transformation allowed real-time process monitoring to be performed. This capability had not previously been accessible on-scale using traditional off-line testing and provided a greater on-scale understanding of the conversion kinetics. In-line testing was less labor intensive, time demanding and also reduced the exposure of the plant operators to the process. Connection of the NIR software to the DCS and Aspentech IP21 process browser allowed remote monitoring of the conversion alongside more conventional physical process data.
For the majority of batches the PAT measurement indicated that the form conversion reached completion several hours prior to the end of processing thus indicating a cycle time reduction may be possible. The replacement of the qualitative, off-line spectroscopic test with an automated in situ method enables cycle time advantages, an increase in process efficiency and real time quality assurance of the finished API. It also offers possible labor savings in the small differences between the PLS model diagnostics were observed between the two manufacturing campaigns and a strategy has been put in place for future model update and maintenance. Application of the PLS model on pilot plant scale, where the conversion was monitored with different equipment (both NIR spectrometer and optical probe) showed that this modeling approach was transferable between NIR equipment and scale.
Acknowledgements
We would like to acknowledge the scientific input of the members of the GSK pazopanib project team. We thank Taniya Mandal for her contributions to analysis of the NIR data. We would also like to acknowledge the members of Technical Development group in Jurong global manufacturing and supply for their hard work on implementing and maintaining the NIR method.References
- Pharmaceutical cGMPs for the 21st Century—a Risk Based Approach, US Department of Health and Human Services, US Food and Drug Administration, September 2004 Search PubMed.
- Guidance for Industry Q8 Pharmaceutical Development, US Department of Health and Human Services, US Food and Drug Administration, May 2006 Search PubMed.
- J. W. Mullin, in Crystallization, Butterworth-Heinemann, Oxford, 3rd edn, 1993 Search PubMed.
- E. L. Paul, H.-H. Tung and M. Midler, Powder Technol., 2005, 150, 133–143 CrossRef CAS.
- R. D. Braatz, Annual Reviews in Control, 2002, 26, 87–99 Search PubMed.
- T. L. Threlfall, Analyst, 1995, 120, 2435–2460 RSC.
- L. X. Yu, R. A. Lionberger, A. S. Raw, R. D'Costa, H. Wu and A. S. Hussain, Adv. Drug Delivery Rev., 2004, 56, 349–369 CrossRef CAS.
- R. Kobayashi, Y. FujiMaki, T. Ukita and Y. Hiyama, Org. Process Res. Dev., 2006, 10, 1219–1226 Search PubMed.
- D. J. Am Ende and M. J. Preigh, Curr. Opin. Drug Discovery Dev., 2000, 3(6), 699–706 CAS.
- A. Caillet, F. Puel and G. Fevotte, Int. J. Pharm., 2006, 307, 201–208 CrossRef CAS.
- M. Birch, S. J. Fussell, P. D. Higginson, N. McDowall and I. Marziano, Org. Process Res. Dev., 2005, 9, 360–364 Search PubMed.
- G. Fevotte, Chem. Eng. Res. Des., 2007, 85(A7), 906–920 CrossRef CAS.
- J. Calderon De Anda, X. Z. Wang, X. Lai, K. J. Roberts, K. H. Jennings, M. J. Wilkinson, D. Watson and D. Roberts, AIChE J., 2005, 51(5), 1406–1414 CrossRef CAS.
- D. D. Dunuwila and K. A. Berglund, J. Cryst. Growth, 1997, 179, 185–193 CrossRef CAS.
- F. Lewiner, J. P. Klien, F. Puel and G. Fevotte, Chem. Eng. Sci., 2001, 56, 2069–2084 CrossRef CAS.
- K. Pollanen, A. Hakkinen, S. P. Reinikainen, J. Rantanen, M. Karjalainen, M. Louhi-Kultanen and L. Nystrom, J. Pharm. Biomed. Anal., 2005, 38, 275–284 CrossRef.
- P. Barrett, B. Smith, J. Worlitschek, V. Bracken, B. O’Sullivan and D. O’Grady, Org. Process Res. Dev., 2005, 9(3), 348–355 Search PubMed.
- J. Scholl, D. Bonalumi, L. Vicum and M. Mazzotti, Cryst. Growth Des., 2006, 6(4), 881–891 CrossRef.
- B. O'Sullivan, P. Barrett, G. Hsiao and A. Carr, Org. Process Res. Dev., 2003, 7, 977–982 Search PubMed.
- L. E. O'Brien, P. Timmins, A. C. Williams and P. York, J. Pharm. Biomed. Anal., 2004, 36(2), 335–340 CrossRef CAS.
- C. Starbuck, A. Spartalis, L. Wai, J. Wang, P. Fernandez, C. M. Lindemann, G. X. Zhou and Z. Ge, Cryst. Growth Des., 2002, 2(6), 515–522 CrossRef CAS.
- T. Vankeirsbilck, A. Vercauteren, W. Baeyens, G. Van der Weken, F. Verpoort, G. Vergote and J. P. Remon, TrAC, Trends Anal. Chem., 2002, 21(12), 869–877 CrossRef CAS.
- X. Yang, J. Lu, X. Wang and C. B. Ching, J. Raman Spectrosc., 2008, 39, 1433–1439 CrossRef CAS.
- K. Kogermann, J. Aaltonen, C. J. Strachan, K. Pollanen, P. Veski, J. Heinamaki, J. Yliruusi and J. Rantanen, J. Pharm. Sci., 2007, 96(7), 1802–1820 CrossRef CAS.
- F. Wang, J. A. Watcher, F. J. Antosz and K. A. Berglund, Org. Process Res. Dev., 2000, 4(5), 391–395 Search PubMed.
- A. D. Patel, P. E. Luner and M. S. Kemper, Int. J. Pharm., 2000, 206, 63–74 CrossRef CAS.
- T. Norris, P. K. Aldridge and S. Sekulic, Analyst, 1997, 122, 549–552 RSC.
- M. Blanco, M. Alcala, J. M. Gonzalez and E. Torras, Anal. Chim. Acta, 2006, 567, 262–268 CrossRef CAS.
- G. Fevotte, J. Calas, F. Puel and C. Hoff, Int. J. Pharm., 2004, 273, 159–169 CrossRef CAS.
- P. K. Aldridge, C. L. Evans, H. W. Ward, II and S. T. Colgan, Anal. Chem., 1996, 68(6), 997–1002 CrossRef CAS.
- M. Blanco, J. Coello, S. Iturriaga, S. Maspoch and C. De la Pezuela, Analyst, 1998, 123, 135R–150R RSC.
- G. Reich, Adv. Drug Delivery Rev., 2005, 57, 1109–1143 CrossRef CAS.
- L. Luypaert, D. L. Massart and Y. Vander Heyden, Talanta, 2007, 72, 865–883 CrossRef CAS.
- Y. Roggo, P. Chalus, L. Maurer, C. Lema-Martinez, A. Edmond and N. Jent, J. Pharm. Biomed. Anal., 2007, 44, 683–700 CrossRef CAS.
- R. N. Feudale, N. A. Woody, H. Tan, A. J. Myles, S. D. Brown and J. Ferre, Chemom. Intell. Lab. Syst., 2002, 64, 181–192 CrossRef CAS.
- E. Bouversesse and D. L. Massart, Chemom. Intell. Lab. Syst., 1996, 32, 201–213 CrossRef CAS.
- A. Andrew and T. Fearn, Chemom. Intell. Lab. Syst., 2004, 72, 51–56 CrossRef CAS.
- E. L. Bergman, H. Brage, M. Josefson, O. Svensson and A. Sparen, J. Pharm. Biomed. Anal., 2006, 41, 89–98 CrossRef CAS.
- M. R. Smith, R. D. Jee, A. C. Moffat, D. R. Rees and N. W. Broad, Analyst, 2004, 129, 806–816 RSC.
- Y. Li, D. Q. Liu, S. Yang, R. Sudini, M. A. McGuire, D. S. Bhanushali and A. S. Kord, J. Pharm. Biomed. Anal., 2010, 52, 493–507 CrossRef CAS.
- C. A. Castaneda and H. L. Gomez, Future Oncol., 2009, 5(9), 1335–1348 Search PubMed.
- R. G. Brereton, in Chemometrics: Data Analysis for the Laboratory and Chemical Plant, John Wiley and Sons Ltd, 2003 Search PubMed.
- A. Savitzky and M. J. E. Golay, Anal. Chem., 1964, 36, 1627–1639 CrossRef CAS.
- L. Nørgaard, A. Saudland, J. Wagner, J. P. Nielsen, L. Munck and S. B. Engelsen, Appl. Spectrosc., 2000, 54, 413–419 CAS.
- R. J. Barnes, M. S. Dhanoa and S. J. Lister, Appl. Spectrosc., 1989, 43, 772–777 CAS.
- G. X. Zhou, X. Ge, J. Dorwart, B. Izzo, J. Kukura, G. Bicker and J. Wyvratt, J. Pharm. Sci., 2003, 92(5), 1058–1064 CrossRef CAS.
- R. A. Forbes, M. L. Persinger and D. R. Smith, J. Pharm. Biomed. Anal., 1996, 15(3), 315–327 CrossRef CAS.
- W. Plugge and C. Van der Vlies, J. Pharm. Biomed. Anal., 1992, 10(11–12), 797–803 CrossRef CAS.
- E. Dreassi, G. Ceramelli, L. Savini, P. Corti, P. L. Perruccio and S. Lonardi, Analyst, 1995, 120, 1005 RSC.
- E. Dreassi, G. Ceramelli, P. Corti, P. L. Perruccio and S. Lonardi, Analyst, 1995, 120, 319 RSC.
- E. Dreassi, G. Ceramelli, P. L. Perruccio and P. Corti, Analyst, 1998, 123, 1259–1264 RSC.
- A. Candolfi and D. L. Massart, Appl. Spectrosc., 2000, 54, 48–53 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |