Separation and quantification of short-chain coenzyme A in plant tissues by capillary electrophoresis with laser-induced fluorescence detection
Yongqing
Jiang
a,
Basil
Nikolau
b and
Yinfa
Ma
*a
aDepartment of Chemistry, Missouri University of Science and Technology, 400 West 11th Street, Rolla, MO, USA. E-mail: yinfa@mst.edu; Fax: +573-341-6033; Tel: +573-341-6220
bDept. of Biochemistry, Biophysics and Molecular Biology, Iowa State University, Ames, IA 50011
First published on 18th October 2010
Abstract
Coenzyme A (CoA) is a group of active metabolic compounds that facilitate over 100 chemical reactions in animal and plant cells. It mainly serves as an acyl carrier in many metabolic reactions and initiates the tricarboxylic acid cycle that produces more than 90% of the energy required for life processes. Measurements of short-chain and long-chain CoA compounds in a variety of tissues by using high-performance liquid chromatography (HPLC) and capillary electrophoresis-ultraviolet (CE-UV) detection have been reported, but these techniques do not allow one to simultaneously determine all the possible coexisting CoAs and their derivatives in plant tissues with sufficient sensitivity. In this paper, a method of quantitative determination of 5 short-chain CoAs in plant tissues by using capillary electrophoresis with laser-induced fluorescence detection (CE-LIF) was developed. Under optimized derivatization and electrophoresis conditions, different CoAs that were derivatized with fluorescein-5-isothiocyanate (FITC) were separated and quantified at the pmole level. A fused silica capillary with a 75 μm (i.d.) × 57 cm was used for the separation and 150 mM borate buffer (pH 9.00) was used as a background electrolyte. The separation was carried out at 25 kV and completed in less than 13 min. The effects of derivatization time, buffer concentrations, and pH values on derivatization efficiency were also systematically investigated. This newly developed CE-LIF method can be used to detect CoAs in both plant and animal tissues.
1. Introduction
Coenzyme A (CoA) is one of the most active metabolic compounds. It facilitates more than 100 chemical reactions in cells, including the metabolism of amino acids, carbohydrates, and lipids.1–3 Most importantly, CoA initiates the tricarboxylic acid (TCA) cycle that produces more than 90% of the energy required for life processes.4 CoA serves primarily as an acyl carrier in many metabolic reactions due to its active thiol (SH) group, which covalently bonds to an acyl group to form thioesters.5,6 The acyl group can then be actively transferred to various acceptor molecules due to its high free energy released during hydrolysis, as in the case of fatty acid biosynthesis. The chemical structure of CoA, and the different portions of the molecule are illustrated in Fig. 1. Research data have also shown that CoA and its derivatives have a site-specific and reversible interaction with certain enzyme systems7 and can act well below the critical micelle level.8 They are considered to be important effectors in cell metabolism because of this unique function. Despite the importance of this molecule in many crucial reactions, it accumulates at low concentrations in cells. In fact its low abundance has been a major hindrance in elucidating the metabolism of this molecule and elucidating metabolic processes in which it is required as a cofactor. To discover the detailed functions of CoA and its derivatives in tissues and subcellular organelles, it is critical to develop an efficient, simple, and sensitive method to detect CoA compounds in biological samples.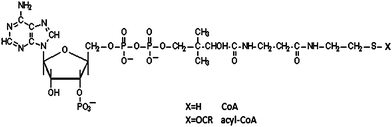 | ||
| Fig. 1 Chemical structure of Coenzyme A. | ||
Different methods have been reported for determining short-chain and long-chain CoA compounds in a variety of tissues, including enzymatic assays,9,10 high- performance liquid chromatography (HPLC) conjugated with either spectroscopic or fluorometric detection and capillary electrophoresis with UV detection.11,12 Since enzymatic assays only allow the determination of one CoA compound at a time, they are not useful for simultaneous determination of all possible coexisting CoAs and their derivatives. Although the HPLC method has long been used as the main technique for measurement of acyl CoA esters, it has quite a few drawbacks, including (1) long running times (45–120 min) associated with generating a large amount of organic wastes, (2) inadequate separation of unknown compounds with short-chain CoAs leading to misidentification and inaccurate quantitation, (3) large shifts of baseline during gradient elution programming, which often requires further solvent purification, and (4) tedious procedures for sample preparation and sample analysis. Capillary electrophoresis (CE) has proven to be a rapid, simple, and sensitive technique for separating charged biomolecules with very high resolution.13–16 However, the sensitivity of the current method using UV as a detector17 cannot meet the requirement of CoA measurement in some biological samples. In this paper, we have demonstrated a capillary electrophoresis-laser induced fluorescence (CE-LIF) method for the first time to separate and quantify CoA compounds in plant leaves. CoA compounds first were derivatized with fluorescein-5-isothiocyanate (FITC), separated by CE and then detected using LIF in order to improve detection sensitivity. This fast, sensitive and reliable method can be applied to determine CoAs in both plant and animal tissues.
2. Experimental
2.1. Chemicals
CoA standards including isovaleryl CoA, acetyl CoA, n-propionyl CoA, isobutyryl CoA and malonyl CoA were purchased from Sigma-Aldrich (St. Louis, MO, USA). FITC was obtained from Invitrogen (Carlsbad, CA, USA). All other chemicals that were used for buffer preparation and capillary rinsing, such as borate, acetone, methanol, sodium hydroxide and hydrochloric acid, were also from Sigma-Aldrich (St. Louis, MO, USA) and of analytical reagent grade unless stated otherwise. Deionized water (18.2 MΩ) from Millipore Simplicity 185 - system (Millipore, MA, USA) was used to prepare standard solutions, background electrolyte (BGE), and other solutions.2.2. Preparation of BGE
The derivertization buffer and BGE solution containing 170 mM and 150 mM borate were prepared respectively with deionized water and the pH were adjusted to 9.00 by adding 1.0 M NaOH in the buffer before diluting it to the final volumes. These solutions were filtered through the 0.45μm nylon membrane filter before use.2.3. Preparation of standard CoA solutions
The 1.0 mM of CoAs and FITC stock solutions were prepared in DI water and acetone respectively. All stock solutions were kept at −20 °C. The working solutions were prepared by diluting the stock solutions to the desired concentrations.2.4. Sample extraction
Cassava leaves were harvested, immediately frozen in liquid N2 and stored at −80 °C until used. As described in the literature,18 before analysis, the frozen leaves (0.5–1.0 g fresh weight) was powdered using mortar and pestle, and then the powder was suspended in 2.0 mL of 5% (w/v) iced trichloroacetic acid (TCA). The TCA suspensions were vortexed, centrifuged at 25,000g for 10 min at 0 °C, and the supernatant was recovered. Repeated partitioning against diethyl ether removed TCA from the extracts, which were then dried under vacuum. The residues were dissolved in DI H2O and ready for derivatization.2.5. Derivatization procedure
Twelve μL CoA working solution or extracts was mixed with 30 μL of 1.0 mmol/L FITC, 258 μL of 0.17 mol/L borate buffer, vortexed and then reacted in the dark at 40 °C in dry incubator for 10 h. Then the 300 μL mixed solution was diluted with 600 μL H2O before injection into the CE system. A blank solution was prepared at the same time to serve as a control.2.6. Instrumentation
All CE experiments were carried out on a Beckman Coulter P/ACE MDQ instrument (Beckman Instruments, Fullerton, CA, USA) equipped with a LIF detector. Excitation was at 488 nm (argon ion laser) and the emission intensity was monitored at 520 nm (band-pass filter, bandwidth 10 nm). Electrophoretic data were acquired and analyzed by 32 Karat software versions 4. Separations were performed in fused silica capillaries (Polymicro Technologies, Phoenix, AZ) with 75μm (i.d.) × 57cm (effective length). New capillaries were conditioned by rinsing with methanol for 15 min, deionized water for 5 min, 1.0 M HCl for 5 min, followed by deionized water for 5 min again, then 1.0 M NaOH for 20 min and deionized water for 5 min. The capillary was rinsed with deionized water for 5 min and then pre-run with BGE for 20 min under 25 kV every morning to obtain the best reproducibility. Samples were injected into the capillary at 0.5 psi for 10 s. After each analysis, the capillary was rinsed successively with 0.1 M NaOH for 1.0 min, deionized water, and BGE for 2.0 min respectively. CoA separation was carried out at 25 kV under 25 °C.3. Results and discussion
CoAs are suitable to conjugate with fluorescence agent FITC and then be analyzed by capillary electrophoresis, due to the amine group in the molecule, the charges they carry, diverse molecular weight, and hydrophilic property of the derivatives. The sensitivity of the CoA measurement in tissue depends on a derivatization efficiency and analytical parameters, such as derivatization agent, running buffer composition, pH, and concentration, the applied voltage on CE, the length and diameter of the capillary, and the sample size introduced, and so on. All these conditions were investigated systematically in this study in order to get the optimum derivatization and separation conditions. Fig. 2 showed the separation of 5 CoA standards under optimal conditions by using CE-LIF. The derivatization was conducted in a 170 mM borate buffer at pH = 9.00 under 40 °C for 10 h in dark. The running buffer was composed of 150 mM borate (pH = 9.00) and the separation was conducted at 25 kV with an LIF detector (excitation wavelength at 488 nm and emission wavelength at 520 nm) at temperature of 25 °C. | ||
| Fig. 2 Electropherogram of the separation of 5 CoA standards under optimal conditions by using CE-LIF. The derivatization was conducted in a 170 mM borate buffer at pH = 9.00 under 40 °C for 10 h in dark. The running buffer was composed of 150 mM borate (pH = 9.00) and the separation voltage was 25 kV with LIF detector. The separation was finished in a capillary with 75μm (i.d.) × 57cm (effective length) at 25 °C. Sample was injected under 0.5psi for 10s. Peak identification: 1, Isovaleryl coenzyme A; 2, Acetyl coenzyme A; 3, n-Propionyl coenzyme A; 4, Isobutyryl coenzyme A; 5, Malonyl coenzyme A. | ||
3.1. Optimization of derivatization
FITC has been widely used in the derivatization of amines in CE,19,20 because it can react with primary and second amines and forms a derivative with an excitation maximum at 488 nm, which is coincident with the argon laser, and an emission maximum at 516 nm. The product information recommended condition for FITC derivatization is 0.2 mol/L in a carbonate buffer. In order to use the same buffer for derivatization and separation, and to avoid the bubble-formation problem of carbonate at the same time, borate was used as derivatization buffer. Eight different borate concentrations (30, 50, 100, 150, 170, 200, 250 and 300 mM) were examined to compare the derivatization efficiency (data was not shown). With the increase of the borate concentrations, the peak intensities were increased gradually until it reached 170 mM. No apparent improvement in derivatization efficiency was observed when borate concentrations were above 170 mM. Since concentrated buffer solution may adversely affect the stability of derivatives,19 170 mM borate was chosen as the derivatization buffer concentration, even though higher concentration buffer may slightly enhance one of the derivatives.The most crucial parameter for the amine derivatization using FITC is the buffer pH. Slightly basic buffer would be beneficial for derivatization,19 since the derivatization involves the deprotonation of the amino-group, and basic solution will help shifting the dissociation equilibration to the deprotonation and therefore improve the derivatization efficiency. Buffer pH effects on derivatization efficiency of five CoAs were investigated and the results were shown in Fig. 3. The results in Fig. 3 demonstrated that the derivatizing pH had remarkably effects on the derivatization efficiency and the efficiency increased continuously as pH increased until about pH 9.0, at which the fluorescence response achieved the highest value. Afterwards, the reaction efficiency decreased as the pH went up. Therefore, an optimized pH for derivatization in this study was maintained at 9.0 for the highest reaction efficiency. The pH of the derivatization buffer not only affected derivatization efficiency, but also affected separation result of capillary electrophoresis. In most cases, a good separation can be obtained when the pH of derivatization buffer is the same as that of the running buffer.19 When the pH of derivatization buffer is lower than that of the running buffer, the acidity of sample plug will result in adsorption of FITC on the capillary wall and the tailing system peak will interfere with analyte peaks. Therefore, the pH of the derivatization buffer was the same as that of the running buffer in this study, which will be discussed later.
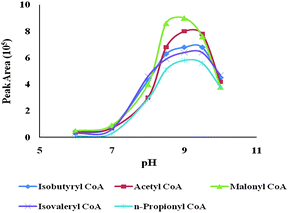 | ||
| Fig. 3 The derivatizing pH effect on the peak areas of 5 CoA standards. The experimental conditions were the same as those of Fig. 2, except for the pHs of the derivatizaion buffer. | ||
FITC to CoA ratio was also investigated by using n-Propionyl CoA (due to the structural similarity of CoAs) and the results were shown in Fig. 4. The efficiency of the derivatization reaction increased with the increasing of the FITC to CoA ratio until the ratio reached to 5 to 1. In addition, higher free FITC concentration caused interference and resulted in poor separation and quantification. Therefore, a FITC to CoA ratio of 5 to 1 was adopted in this study.
 | ||
| Fig. 4 Optimization of FITC to CoA ratio. The experimental conditions were the same as those of Fig. 2, except for the FITC to CoA ratio. Peak identifications were the same as those in Fig. 2. | ||
The temperature and time of derivatization reaction were also found to affect the derivatization efficiency. After a series of experiments (data was not shown), 40 °C was considered being the best temperature, because reaction went faster at a relatively higher temperature. However, it was found that the derivatization product will decompose at a temperature higher than 40 °C. The systematic study of the derivatization time effect on the derivatization efficiency was shown in Fig. 5. Based on the results in Fig. 5, 10 h derivatization time was chosen prior to injection to CE for analysis. Longer reaction time may induce product decomposition.
 | ||
| Fig. 5 The derivatizaion time effect on the separation of 5 CoA standards. The experimental conditions were the same as those of Fig. 2, except for the time of the derivatizaion. Peak identifications were the same as those in Fig. 2. | ||
3.2. Optimization of separation
Running buffer is the key media for CE separation and it must be optimized to achieve a good separation. A good running buffer should have the following characteristics: good resolution of sample molecules, low current and low Joule heat, high fluorescence quantum yield, and good buffer pH range and capacity. Borate solution has been chosen as the running buffer in this study because of its low current and good pH range for FITC derivatives. As mentioned above, the derivatization reaction was conducted in the alkaline solution.The pH of the running buffer had great influence on the fluorescence intensities of FITC derivatives. According to the FITC product information,21 the fluorescence intensities are low and steady when pH is below 3.0. With the increasing of pH from 3.0 to 6.0, the fluorescence intensities rapidly increase. When pH is higher than 6.0, the fluorescence intensities reaches to a maximum and become steady. Therefore, good detection sensitivity and signal stability can be obtained under a basic condition. For the current study, four pHs (8.50, 9.00, 9.50 and 10.00) were tested. The optimal sensitivity and separation was obtained under pH 9.00, which coincide with the previous discussion. The retention time became longer at a higher pH. Separation efficiency was also greatly affected by the BGE concentration. Since FITC-CoA derivatives were negatively charged and migrated against the electroosmotic flow (EOF), a low molar concentration of BGE buffer, which decreased the EOF, decreased the separation efficiency. When the BGE concentration was above 150 mM, the EOF and Joule heating greatly increased, therefore causing a decrease in separation efficiency also. Based on the experimental results (data not shown), 150 mM borate was used as BGE.
The effect of applied voltages on the separation of five analytes was also investigated (data not shown). It was found that with the increases of the applied voltage, the analytes migration times were decreased and fast separation was obtained. However, when the applied voltage was higher than 25 kV, the analyte peaks were too close and it is hard to observe a baseline separation. In addition, due to the Joule heat caused by the increased electrophoretic current, the peak width expanded, which reduced the separation efficiency. Therefore, 25 kV was selected as the optimized applied voltage in our study.
3.3. Stability
Time and temperature are critical to the stability of the CoA metabolites.18 The analyte stabilities before and after derivatizaion were assessed (data was not shown). Freeze-thaw cycles was not recommended in Co-A analysis by using this method. It was also found that the derivatives were stable for at least one week in the refrigerator (4 °C). The derivatives should be analyzed right after the derivatization, otherwise stored at 4 °C. At room temperature, the derivatives were only stable for 5 h. A longer storage time may cause product decomposition.3.4. Linearity, detection limits, and reproducibility
As shown in Fig. 2, 5 FITC-CoA derivatives and unreacted labeling agents were well separated, demonstrating the excellent separation of these analytes under the optimized conditions. Five CoAs, which commonly exist in biological extracts and are of interest to biological researchers, were completely separated in less than 13 min. The migration order was 1, Isovaleryl coenzyme A; FITC; 2, Acetyl coenzyme A; 3, n-Propionyl coenzyme A; 4, Isobutyryl coenzyme A; 5, Malonyl coenzyme A. This elution order can be explained on the basis of the molecular structure and mass-to-charge ratio. The reason of the position and 2 peaks for isovalery CoA was unknown and will be further studied. For all 5 FITC-CoAs, they all moved reasonably fast toward to the detector side due to their high mass-to-charge ratio.The linearity, reproducibility and detection limits for determination of FITC- CoA derivatives were listed in Table 1.
| CoA Standards | RSD (%) | Linearity (R2) | Detection Limit (nM, S/N = 3) | |
|---|---|---|---|---|
| Migration Time | Peak Area | |||
| Acetyl coenzyme A | 2.27 | 3.38 | 0.9986 | 1.19 |
| Malonyl coenzyme A | 1.97 | 2.86 | 0.9972 | 0.26 |
| Isobutyryl coenzyme A | 2.06 | 3.05 | 0.9988 | 1.37 |
| n-Propionyl coenzyme A | 2.13 | 3.32 | 0.9968 | 0.12 |
| Isovaleryl coenzyme A | 3.89 | 5.46 | 0.9932 | 0.61 |
The reproducibility was expressed as relative standard deviation (RSD) values for both migration time and peak area and was calculated by using 0.1 mmol/L CoA standard solutions (n = 5). The RSD values of migration time were between 1.97% and 3.89% and the RSD for peak areas were between 2.86% and 5.46%, which indicates a good reproducibility. The linearity of the method was determined by using standard mixtures at six concentrations from 0.5 to 500 nmol/L and correlation coefficients ranged from 0.9932 to 0.9988. Detection limits varied from 0.12 and 1.37 nmol/L with a signal to- noise ratio (S/N) of 3, which was sensitive enough to detect tissue extract CoAs.
3.5. Analysis of coenzyme A's in plant leave cell extracts
Plant leaves were analyzed (n = 5) using the optimized CE-LIF method. A representative electropherogram of the separation of interest analytes from plant extracts was shown in Fig. 6. CoA peaks in the cell extracts were identified by retention times and standard additions. The CoA extraction recoveries in leave extracts and the CoA levels obtained from the analysis of leave extracts were shown in Table 2. The results were comparable to those of HPLC methods for CoAs in plant tissue.18 | ||
| Fig. 6 Representative electropherogram of the separation of 5 CoAs from plant extracts under optimal conditions by using CE-LIF. The experimental conditions and peak identifications were the same as those of Fig. 2. | ||
| CoAs | Concentration (nmol/g fresh weight) | Recovery (%) ± SD |
|---|---|---|
| Acetyl coenzyme A | 5.19 ± 0.2 | 87.6 ± 1.64 |
| Malonyl coenzyme A | 0.96 ± 0.1 | 83.5 ± 2.21 |
| Isobutyryl coenzyme A | 3.55 ± 0.05 | 90.8 ± 3.22 |
| n-Propionyl coenzyme A | 0.92 ± 0.01 | 88.7 ± 1.58 |
| Isovaleryl coenzyme A | 2.01 ± 0.05 | 91.2 ± 3.16 |
4. Conclusions
A sensitive CE-LIF method was successfully developed for determination of selected CoAs in plant cell extracts. FITC was used for the off-line derivatization and it was found that the derivatization efficiency was the highest in 170 mM borate buffer at pH 9.00. By using 150 mM borate BGE at pH 9.0, FITC-CoAs were well separated in less than 13 min at a voltage of 25 kV. This CE-LIF method is sensitive, fast, and environment friendly. It can be applied to measure CoAs in both plant and animal tissue and has the potential to be used for metabolite study in which CoAs are required.Acknowledgements
This research was supported by Plant Sciences Institute at Iowa State University and startup funds awarded to Dr Yinfa Ma from Missouri University of Science and Technology. The authors thank them for their financial support. The authors also thank Dr Tahzeeba Hossain at the Donald Danforth Plant Science Center for providing the plant leaves.References
- I. A. Graham and P. J. Eastmond, Prog. Lipid Res., 2002, 41, 156–181 CrossRef CAS.
- J. Heider, FEBS Lett., 2001, 509, 345–349 CrossRef CAS.
- M. C. Hunt, K. Solaas, B. F. Kase and S. E. H. Alexson, J. Biol. Chem., 2002, 277, 1128–1138 CrossRef CAS.
- A.Y., Metabolism of Coenzyme A, 1975, 1–25 Search PubMed.
- M. Mancha, G. B. Stokes and P. K. Stumpf, Anal. Biochem., 1975, 68, 600–608 CrossRef CAS.
- D. V. Voet, J.G. and C. W. Pratt, Fundamentals of Biochemistry, 1999 Search PubMed.
- A. Shug, E. Lerner, C. Elson and E. Shrago, Biochem. Biophys. Res. Commun., 1971, 43, 557–563 CrossRef CAS.
- P. S. Tippett and K. E. Neet, J. Biol. Chem., 1982, 257, 12839–12845 CAS.
- D. Veloso and R. L. Veech, Anal. Biochem., 1974, 62, 449–460 CrossRef CAS.
- D. Veloso and R. L. Veech, Methods Enzymol., 1975, 35, 273–278 Search PubMed.
- T. R. Larson and I. A. Graham, Plant J., 2001, 25, 115–125 CrossRef CAS.
- T. R. Larson and I. A. Graham, Biochem. Soc. Trans., 2000, 28, 575–577 CrossRef CAS.
- M. Girod and D. W. Armstrong, Electrophoresis, 2002, 23, 2048–2056 CrossRef CAS.
- L. Kotler, H. He, A. W. Miller and B. L. Karger, Electrophoresis, 2002, 23, 3062–3070 CrossRef CAS.
- W. Wei and E. S. Yeung, Anal. Chem., 2002, 74, 3899–3905 CrossRef CAS.
- Y. Jiang and Y. Ma, Anal. Chem., 2009, 81, 6474–6480 CrossRef CAS.
- G. Liu, J. Chen, P. Che and Y. Ma, Anal. Chem., 2003, 75, 78–82 CrossRef CAS.
- A. W. Tumaney, J. B. Ohlrogge and M. Pollard, J. Plant Physiol., 2004, 161, 485–488 CrossRef CAS.
- S. Xiong, H. Han, R. Zhao, Y. Chen and G. Liu, Biomed. Chromatogr., 2001, 15, 83–88 CrossRef CAS.
- J. Mattusch, G. Huhn and R. Fresenius Wennrich, Fresenius J. Anal. Chem., 1995, 351, 732–738 CAS.
- Molecular Probes, The Handbook-A Guide to Fluorescent Probes and Labeling Technologies, website edition, 2010 Search PubMed.
| This journal is © The Royal Society of Chemistry 2010 |