The latent ampholytic nature of glycosaminoglycan (GAG) oligosaccharides facilitates their separation by isoelectric focusing†
Joseph
Holman‡
,
Mark Andrew
Skidmore‡
,
Timothy Robert
Rudd
and
Edwin Alexander
Yates
*
The University of Liverpool, Crown Street, Liverpool, UK. E-mail: eayates@liv.ac.uk; Fax: +44 (0)151 795 4404; Tel: +44 (0)151 795 4429
First published on 3rd September 2010
Abstract
Glycosaminoglycans are medically and biologically important polysaccharides. Glycosaminoglycan oligosaccharides are of increasing pharmaceutical relevance but, oligosaccharides from two in particular, heparan sulfate and heparin, are difficult to separate owing to their structural complexity. This hinders further progress because it is difficult to obtain pure saccharides in sufficient quantities. The ampholytic nature of heparin and heparan sulfate is realised by reversible de-N-sulfation or de-N-acetylation to reveal free amine groups, providing an opportunity to separate them using isoelectric focusing. Ampholytic heparan sulfate and heparin saccharides are characterised by low pI values, and isoelectric focusing required bespoke polyacrylamide gels capable of supporting pH gradients below pH 2. Representative oligosaccharide pools from heparin and chemically modified heparin, which had been fractionated previously to the best current levels by high performance anion exchange chromatography and gel permeation chromatography, were further separated and focused. The isoelectric focusing method provides an additional separation dimension for glycosaminoglycan oligosaccharides and has implications for the future development of related semi-preparative techniques.
Introduction
The inherent structural complexity of the biologically and medically important anionic glycosaminoglycan (GAG) polysaccharides is a major obstacle to the elucidation of their structure–function relationships, which remains a major challenge in the field. This complexity is due to their non-template driven biosynthesis, involving the action of several enzymes, producing linear chains that can contain isomeric sequences. The GAG family of carbohydrates includes hyaluronic acid (HA), chondroitin (CS-A, CS-C) and dermatan sulfate (CS-B or DS), keratan sulfate (KS), heparin and heparan sulfate (HS). Repeating disaccharide subunits form the backbone of all GAGs, with varying degrees of N-acetylation and O-sulfation, with the exception of HA, which is not sulfated. The two closely related GAGs, heparin and HS share underlying constituent disaccharide repeats (Fig. 1) and are undoubtedly the most complex family members but, differ primarily from other GAGs in having a mixed uronic acid content (β-D-GlcA and its epimer α-L-IdoA) and uniquely, a degree of N-sulfation. Heparin and its derivatives are widely used as convenient proxies for HS, possessing many of its biological activities.1–4 Heparan sulfate, in contrast to heparin (found exclusively within mast cells), is expressed on almost all cell-surfaces in the extracellular matrix, and is characterised by high sequence diversity5 along with a defined domain structure. How, and to what extent, these sequences encode specificity in HS and heparin is at present contentious,5,6 although it has recently been hypothesised that a degree of conformational redundancy could exist and the structural basis of this redundancy has been explored.7 This could also explain the difficulty of finding a simple technique capable of separating HS/heparin oligosaccharides.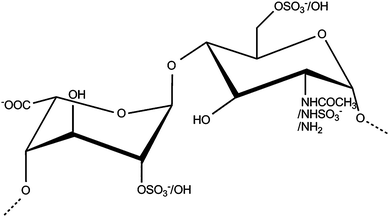 | ||
| Fig. 1 Repeating disaccharide subunit of heparin and heparan sulfate. | ||
Currently, the study of structure–function relationships for this class of carbohydrates relies on the partial depolymerisation of polysaccharide chains to liberate constituent oligosaccharide fragments. Existing sequencing strategies can only examine fragments of modest length,8–10 not intact polysaccharides. Owing to increased interest in GAGs as potential therapeutic agents, including anti-virals,11 anti-inflammatory agents,12,13 secretase inhibitors including the Alzheimer's β secretase BACE-1,1 and disruptors of the rosetting phenomenon in severe malaria,3 there is a pressing need for pioneering structure–function studies. It is sobering to note that it remains a significant technical challenge to isolate fragments of HS or heparin to chromatographic purity, which is a pre-requisite for practically all such sequencing studies.8–10
The separation of HS and/or heparin oligosaccharides at present relies on two well-established techniques: gel permeation chromatography (GPC)14–16 and high-performance anion exchange chromatography (HPAEC).17 Gel permeation chromatography provides a general separation, yielding pools of oligosaccharides with similar hydrodynamic volume, which are often (inaccurately) assumed to possess the same degree of polymerisation. Separation by HPAEC is limited practically by the ability of mechanical pumps to control the salt gradient used to elute the saccharides from the column and by heterogeneities on the surfaces of the cationic beads. These are compounded by possible redundancy between interactions of the anionic sugar structures and the cationic surface of the column matrix. Gel and capillary electrophoresis, on the other hand, have both been employed analytically to separate these sugars on the basis of their overall charge, conformation and hydrodynamic properties10,18–22 and it has been demonstrated that resolution can be improved with chiral additives and other reagents.23 However, only limited resolution has been achieved by any of these methods and the challenge is to find, or generate, physico-chemical properties and complementary methodologies that might provide additional separations. The obstacle of separating saccharides to purity currently restricts the analysis of structure–function relationships in the study of heparin and HS, rather than an inability to sequence per se. This hinders further progress in the pursuit of structure–function relationships, by conventional means8,24 and hampers the construction and subsequent validation of surface-based microarrays6,25,26 ultimately stalling the development of pharmaceutical agents based on these materials.12
Experimental
Materials
All chemicals and solvents were purchased from Sigma-Aldrich (Dorset, UK) except hydrochloric, nitric and trifluoroacetic acids from BDH Laboratory Supplies (Dorset, UK); heptafluorobutyric acid from Rathburn Chemicals Ltd (Walkerburn, UK) and bromoacetic, chloroacetic, chlorodifluoroacetic, dibromoacetic, dichloroacetic, difluoroacetic, hypophosphorous, malonic, oxalic, phosphoric, pyruvic, and tribromoacetic acids from Alfa Aesar (Heysham, UK). HPLC grade water and an InLab® surface pH electrode were obtained from Fisher Scientific (Loughborough, UK). Tinzaparin (Innohep®) was obtained from Leo Pharma (Princes Risborough, UK). Novex IEF pH 3–7 sample buffer was obtained from Invitrogen (Paisley, UK). Cleangel® IEF gels, Cleangel® IEF electrode strips and G-10 columns were purchased from GE Healthcare (Chalfont, UK). Propac-PA1 semi-preparative HPLC-SAX columns were obtained from Dionex (Camberley, UK). TSK gel columns were obtained from Supelco (Dorset, UK). DVC camera and associated software were obtained from the DVC Company Inc. (Austin, Texas, USA).De-N-sulfation
De-N-sulfation was performed essentially as described previously,27,30 with slight modifications. Briefly, tinzaparin, a low molecular weight heparin, was exchanged into the acid form (cation exchange resin) before neutralization with pyridine and freeze-dried to yield the pyridinium salt. De-N-sulfation was achieved by reaction at 65 °C (90 minutes) in anhydrous dimethyl sulfoxide containing 10% anhydrous methanol (v/v). The reaction was cooled to 4 °C using ice and the pH was adjusted with sodium hydroxide (1 M) to pH 9.0, prior to precipitation in ice cold ethanol saturated with sodium acetate. The precipitate was filtered and washed with cold ethanol before resuspension in HPLC grade water. Contaminating salts were removed by dialysis (HPLC grade water). The product was concentrated by rotary evaporation and converted to the acid form (ion exchange resin).Strong anion exchange chromatography
De-N-sulfated oligosaccharide preparations were separated into charge fractions using high performance strong anion exchange chromatography. Samples (20 mg) were injected (1 ml loop) in HPLC grade water onto a Propac PA1 semi-preparative column (9 mm ID, 250 mm length) prior to linear gradient elution with sodium chloride (0–2 M) over 90 minutes. Residual buffer salts were removed using a G-10 de-salting (GE Healthcare) column as per the manufacturer's instructions.Gel filtration chromatography
Charge separated oligosaccharides were further fractionated using gel filtration high performance liquid chromatography. Oligosaccharide samples (200 µg) were injected (20 µl loop) in ammonium acetate (100 mM) before isocratic elution in ammonium acetate (100 mM) over 60 minutes using TSK gel G3000SWXL and TSK G2000SWXL columns connected in series, as previously described by Mulloy et al.15 Eluting peaks were collected and serially lyophilised to remove residual buffer salts.pH gradient gel preparation
A pre-dried Cleangel® was cut into strips (10 × 110 × 0.5 mm). Rehydration of the strip was carried out (1 h) in a mixture of the following acids: bromoacetic, chloroacetic, chlorodifluoroacetic, dibromoacetic, dichloroacetic, difluoroacetic, heptafluorobutyric, hydrochloric, hypophosphorous, maleic, malonic, nitric, oxalic, phosphoric, pyrophosphoric, pyruvic, tribromoacetic, trichloroacetic, and trifluoroacetic, all at 2 mM. Ethylene glycol was added to the rehydration solution (5%, v/v) to reduce electroendosmosis and reduce the level of extruded water on the gel surface.Isoelectric focusing
Samples (50 µg) for IEF were blotted (10 µl) onto a pre-cut electrode strip (5 mm × 10 mm) placed onto the upper gel surface near the cathodic extremity of the gel. The gel was totally immersed in mineral oil to prevent evaporation (Fig. 2). Isoelectric separation was carried out using a 0.4 M hydrochloric acid dampened electrode wick at the anode and a 0.5 M 2-(N-morpholino)ethanesulfonic acid dampened electrode wick at the cathode. The gel was run (180 minutes, 500 V, followed by 180 minutes, 1000 V); the current was limited to a maximum 1 mA for both periods.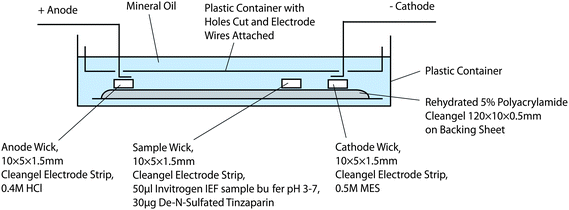 | ||
| Fig. 2 Schematic representation of IEF apparatus. | ||
Determination of pH gradient
The pH across the gel was determined at 10 mm intervals from the anode towards the cathode using an InLab® surface pH electrode and all readings were conducted in triplicate. The electrode was calibrated against known pH reference solutions.Detection of sugars
Focussed saccharides were detected by immersion of the polyacrylamide gels in Azure A (2.7 mM, 10 minutes) before extensive washing in HPLC grade water to remove non-specific background staining. Gels were visualised and recorded using a visible light transilluminator and a DVC 1310C camera coupled with DVC view software (DVC Company Inc.).Results and discussion
The latent amphoteric properties of GAG oligosaccharides are revealed following the selective27,28 and reversible29,30 removal of their N-sulfate or N-acetyl groups (Fig. 3, S1–S3 and S5†) to provide an additional physico-chemical property for their separation. Representative oligosaccharides were first fractionated by the best currently available means, using HPEAC and GPC (in that order, to match that used industrially, in which GPC is an expensive, polishing step) and further separated during isoelectric focusing (IEF) (Fig. 4A and B).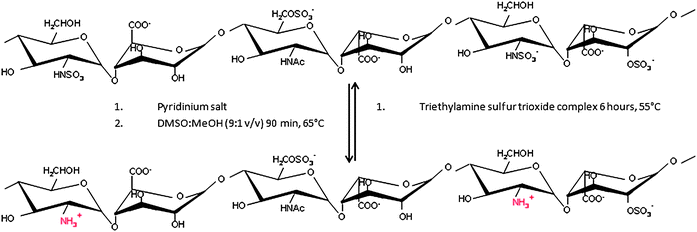 | ||
| Fig. 3 Schematic representation of de-N-sulfation. | ||
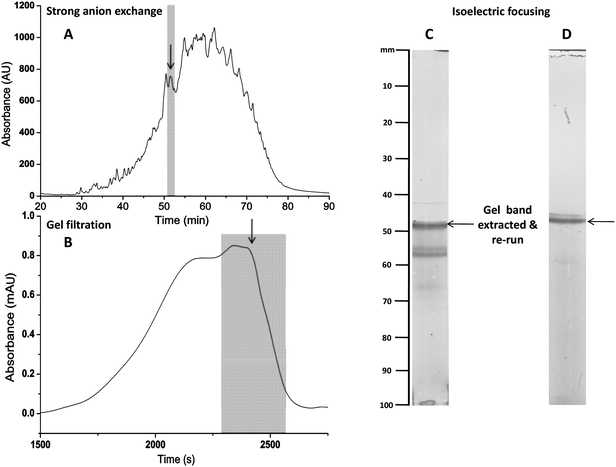 | ||
| Fig. 4 Following fractionation with HPAEC (A) and GPC (B), the de-N-sulfated oligosaccharides from low molecular weight heparin were further separated by isoelectric focusing (IEF) (C). The highlighted band from (C) was cutout, extracted and subjected to IEF for a second time (D). | ||
An earlier exploration of the size-range of polymeric heparin preparations was made by Johnson and Mulloy,14 which relied on conversion into the de-N-sulfated derivative and separation on a polyacrylamide–agarose gel. This followed earlier work by Nader et al.,31 employing unmodified heparin and reporting the apparently paradoxical focusing of these anionic, but not normally ampholytic, sugars on pH gradient polyacrylamide gels. A likely explanation for this behaviour is that the acidic sugars complexed cationic counterions from the carrier ampholytes (presumably with multiple positive charges) imparting the ability to be focused, at least approximately, on pH gradient gels.32–34 However, the banding patterns obtained using the commercial ampholytes,35–38 Pharmalyte® or Ampholine® were similar, regardless of whether or not the heparin sample was de-N-sulfated,14 indicating that the banding did not represent separation, rather, distribution.
The approach described here is distinct from these earlier methods, in that it deliberately generates amphoteric GAG species, by either selective (and reversible) de-N-sulfation, or de-N-acetylation, providing an additional dimension for their differentiation and achieved separation by employing bespoke low pH gradient gels. This provides an additional separation dimension and generates distinct focused patterns for different samples (Fig. 4A and B and 5). The excision of a band and its re-focusing on a second IEF run confirms that IEF has occurred (Fig. 4C and D). The method applies to oligosaccharides derived from heparin, HS and their chemically modified derivatives and exploits the ability to form a stable pH gradient gel at low pH (pH 1–3), using acids with the desired dissociation constants (for pH gradient details see Fig. S4†). Selection of an appropriate set of acids permitted gels to be made conveniently, accessing lower pH ranges than commercially available and facilitated IEF of these highly acidic saccharides. The polyacrylamide gel used (5% T, 3% C) has an open pore structure preventing molecular sieving; gels in excess of 20–25% acrylamide are required to achieve this. IEF provides an additional dimension for the separation of acidic species including HS and heparin oligosaccharides. On a small scale, extraction of the sugars and subsequent re N-sulfation (or re N-acetylation) can be achieved29,30 and the success of this method suggests that other separation techniques exploiting the amphoteric nature of de-N-sulfated and de-N-acetylated oligosaccharides may also be feasible.
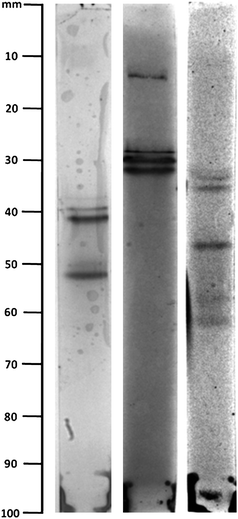 | ||
| Fig. 5 IEF separation of three distinct heparin oligosaccharide pools (de-N-sulfated low molecular weight heparin), post-HPLC fractionation by size and charge chromatographies. | ||
IEF is relatively rapid (∼6 h runs), can deal with ∼50 µg per run (one strip) and, using a simple modification (increasing the dimensions of the supporting structure) of the current set-up, could allow ∼500 µg to be processed. Additional scale-up will be achievable simply by increasing the dimensions of the supporting structure (plastic container in Fig. 2) and the size of the IEF strips. The products of separation containing free amine groups can be directly returned to their original state (i.e. either N-sulfated or N-acetylated), or converted to new products using the complementary reaction (i.e.N-acetylation or N-sulfation).
The isomeric structures of many HS/heparin oligosaccharides render their separation by conventional means a challenge which currently cannot be met. The IEF approach introduces a new dimension of separation hitherto inaccessible and provides the highest level of separation yet achieved. Resolution could be improved by increasing the length of the strips, which works by extending the pH gradient and would require no additional equipment. The technique may also be amenable to conversion into a column format, to increase sample loading.
IEF offers a fundamental improvement in the separation of HS and heparin oligosaccharides and will enable previously inaccessible structure–function experiments to be undertaken.
Acknowledgements
The authors would like to thank the BBSRC and the Welcome Trust for their financial support afforded by project grants BB/G00059X/1 and 082502/Z/07/Z respectively. C. Cosentino of Istituto di Chimica e Biochimica, Milan is thanked for NMR analysis.References
- S. J. Patey, E. A. Edwards, E. A. Yates and J. E. Turnbull, J. Med. Chem., 2006, 49, 6129–6132 CrossRef CAS.
- T. R. Rudd, S. E. Guimond, M. A. Skidmore, L. Duchesne, M. Guerrini, G. Torri, C. Cosentino, A. Brown, D. T. Clarke, J. E. Turnbull, D. G. Fernig and E. A. Yates, Glycobiology, 2007, 17, 983–993 CrossRef CAS.
- M. A. Skidmore, A. F. Dumax-Vorzet, S. E. Guimond, T. R. Rudd, E. A. Edwards, J. E. Turnbull, A. G. Craig and E. A. Yates, J. Med. Chem., 2008, 51, 1453–1458 CrossRef CAS.
- E. A. Yates, S. E. Guimond and J. E. Turnbull, J. Med. Chem., 2004, 47, 277–280 CrossRef CAS.
- B. Casu, Haemostasis, 1990, 20, 62–73 Search PubMed.
- A. K. Powell, E. A. Yates, D. G. Fernig and J. E. Turnbull, Glycobiology, 2004, 14, 17R–30R CrossRef CAS.
- T. R. Rudd and E. A. Yates, Mol. BioSyst., 2010, 6, 902–908 RSC.
- M. Guerrini, T. Agulles, A. Bisio, M. Hricovini, L. Lay, A. Naggi, L. Poletti, L. Sturiale, G. Torri and B. Casu, Biochem. Biophys. Res. Commun., 2002, 292, 222–230 CrossRef CAS.
- C. L. Merry, M. Lyon, J. A. Deakin, J. J. Hopwood and J. T. Gallagher, J. Biol. Chem., 1999, 274, 18455–18462 CrossRef CAS.
- J. E. Turnbull, J. J. Hopwood and J. T. Gallagher, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 2698–2703 CrossRef CAS.
- D. Xu, V. Tiwari, G. Xia, C. Clement, D. Shukla and J. Liu, Biochem. J., 2005, 385, 451–459 CrossRef CAS.
- R. Lever and C. P. Page, Nat. Rev. Drug Discovery, 2002, 1, 140–148 CrossRef CAS.
- C. R. Parish, Nat. Rev. Immunol., 2006, 6, 633–643 CrossRef CAS.
- E. A. Johnson and B. Mulloy, Carbohydr. Res., 1976, 51, 119–127 CrossRef CAS.
- B. Mulloy, C. Gee, S. F. Wheeler, R. Wait, E. Gray and T. W. Barrowcliffe, Thromb. Haemostasis, 1997, 77, 668–674 Search PubMed.
- A. Ziegler and J. Zaia, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2006, 837, 76–86 CrossRef CAS.
- P. A. J. Mourier and C. Viskov, Anal. Biochem., 2004, 332, 299–313 CrossRef CAS.
- J. B. L. Damm and G. T. Overklift, J. Chromatogr., A, 1994, 678, 151–165 CrossRef CAS.
- J. B. L. Damm, G. T. Overklift, B. W. M. Vermeulen, C. F. Fluitsma and G. W. K. Vandedem, J. Chromatogr., 1992, 608, 297–309 CrossRef CAS.
- U. R. Desai, H. M. Wang, S. A. Ampofo and R. J. Linhardt, Anal. Biochem., 1993, 213, 120–127 CrossRef CAS.
- V. Duchemin, I. Le Potier, C. Troubat, D. Ferrier and M. Taverna, Biomed. Chromatogr., 2002, 16, 127–133 CrossRef CAS.
- M. Y. Kim, A. Varenne, R. Daniel and P. Gareil, J. Sep. Sci., 2003, 26, 1154–1162 CrossRef CAS.
- V. Solinova, V. Kasicka, P. Sazelova, T. Barth and I. Miksik, J. Chromatogr., A, 2007, 1155, 146–153 CrossRef CAS.
- B. Tissot, N. Gasiunas, A. K. Powell, Y. Ahmed, Z. L. Zhi, S. M. Haslam, H. R. Morris, J. E. Turnbull, J. T. Gallagher and A. Dell, Glycobiology, 2007, 17, 972–982 CrossRef CAS.
- J. L. de Paz, T. Horlacher and P. H. Seeberger, Methods Enzymol., 2006, 415, 269–292 CAS.
- E. A. Yates, M. O. Jones, C. E. Clarke, A. K. Powell, S. R. Johnson, A. Porch, P. P. Edwards and J. E. Turnbull, J. Mater. Chem., 2003, 13, 2061–2063 RSC.
- Y. Inoue and K. Nagasawa, Carbohydr. Res., 1976, 46, 87–95 CrossRef CAS.
- P. N. Shaklee and H. E. Conrad, Biochem. J., 1984, 217, 187–197 CAS.
- A. G. Lloyd, G. Embery and L. J. Fowler, Biochem. Pharmacol., 1971, 20, 637–648 CrossRef CAS.
- E. A. Yates, F. Santini, M. Guerrini, A. Naggi, G. Torri and B. Casu, Carbohydr. Res., 1996, 294, 15–27 CAS.
- H. B. Nader, N. M. McDuffie and C. P. Dietrich, Biochem. Biophys. Res. Commun., 1974, 57, 488–493 CrossRef CAS.
- E. Gianazza and P. G. Righetti, Biochim. Biophys. Acta, 1978, 540, 357–364 CrossRef CAS.
- P. G. Righetti, R. P. Brown and A. L. Stone, Biochim. Biophys. Acta, 1978, 542, 232–244 CrossRef CAS.
- P. G. Righetti and E. Gianazza, Biochim. Biophys. Acta, 1978, 532, 137–146 CAS.
- H. Engelhardt and M. Martin, Adv. Polym. Sci., 2004, 165, 211–247 CAS.
- C. Simo, M. E. Mendieta, P. Antonioli, R. Sebastiano, A. Citterio, A. Cifuentes and P. G. Righetti, Electrophoresis, 2007, 28, 715–723 CrossRef CAS.
- H. Svensson, Acta Chem. Scand., 1961, 15, 325–341 CAS.
- H. Svensson, Acta Chem. Scand., 1962, 16, 456–466 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: S1—13C NMR spectra of de-N-acetylated porcine mucosal heparin; S2—13C NMR spectra of de-N-acetylated chondroitin sulfate A; S3—13C NMR spectra of de-N-sulfated porcine mucosal low molecular weight heparin; S4—pH gradient of a representative focussed IEF gel; S5—schematic representation of de-N-acetylation. See DOI: 10.1039/c0ay00340a |
| ‡ Authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2010 |