Rapid structure determination of microgram-level drug metabolites using HPLC-MS, fraction collection and NMR spectroscopy
Kim A.
Johnson
,
Xiaohong
Liu
,
Stella
Huang
,
Vikram
Roongta
,
W. Griffith
Humphreys
and
Yue-Zhong
Shu†
*
Pharmaceutical Candidate Optimization, Bristol-Myers Squibb Research & Development, 5 Research Parkway, Wallingford, CT 06492, USA. E-mail: yuezhong.shu@bms.com
First published on 23rd August 2010
Abstract
A robust method for in vitro metabolite generation and facile sample preparation on analytical HPLC was established for rapid structure determination of microgram-level drug metabolites by using high-field NMR equipped with a cryoprobe. A single 1–5 mL incubation of drug candidate (10–30 μM) in microsomes, hepatocytes, or recombinant drug-metabolising enzymes, typically cytochrome P450s and UDP-glucuronosyltransferases, was used for metabolite formation. Following precipitation of proteins and solvent removal, metabolite mixtures were chromatographed with 5–10 injections onto an HPLC-MS system. Metabolites were collected into a 96-well plate, dried, and reconstituted in deuterated NMR solvents. NMR spectra of isolated metabolites were acquired on a 500 MHz spectrometer equipped with a 5 mm cryogenic probe. The methodology has been successfully employed as an extension of HPLC-MS/MS-based metabolite identification and applied frequently to 0.5–10 μg quantities of metabolite. Most structure determinations were achieved rapidly by 1D 1H NMR with satisfactory signal-to-noise ratios, whereas some required 2D NMR data analysis. This report describes the method development and metabolite structure determination using the model compound trazodone. In addition to trazodone, a large number of examples from our laboratories have proven that the microgram-level NMR method avoids time-consuming preparative-scale metabolite generation and purification and circumvents technical complications associated with online LC-NMR. Most importantly, the turnaround time of metabolite structure determination for metabolically unstable compounds using the present methodology is more in sync with the cycle time during which medicinal chemists modify metabolic softspots while performing other iterative lead optimisation activities, demonstrating a real impact on the drug-discovery process.
Introduction
A major aim in drug discovery is the identification of safe and efficacious medicines that can be administered according to convenient once- or twice-daily dosing regimens. Since metabolic processes influence many other parameters that are relevant in this regard, including bioavailability, systemic clearance and toxicology, issues relating to drug metabolism are important in the selection of viable drug candidates. As metabolism-related liabilities, such as metabolic softspots, reactive and potentially toxic metabolites, continue to be a major cause of attrition for drug candidates, biotransformation studies are becoming increasingly important in guiding the refinement of a lead series during drug discovery and in characterising lead candidates prior to clinical evaluation.1 The fast-paced drug discovery process requires biotransformation scientists to identify drug metabolites efficiently in sync with the speed at which medicinal chemists synthesise drug candidates during iterative cycles of lead-optimising activities.2,3 High performance liquid chromatography (HPLC) coupled with tandem mass spectrometry (MS) is widely used as the front-line analytical tool for metabolite identification and the fragmentation of molecules of interest in tandem MS by collision-induced dissociation (CID) generally enables the assignment of Markush-type structures, which may be useful in some instances to assist with drug design effort of medicinal chemistry. However, the product ion spectra are often not informative enough for localising the site of metabolic change, particularly for unknown and unpredicted drug metabolites. In addition, LC-MS has inherent limitations in elucidating regio- and stereo-chemistry. As a tool for structure elucidation, nuclear magnetic resonance (NMR) spectroscopy offers details on atom connectivities within a molecule, and is particularly useful when the exact site of metabolism is of interest for structural modification, but cannot be readily assigned by MS. The application of NMR for metabolite structure determination was a subject of comprehensive review in a recent monograph, where relevant hardware, key parameters and methodology, and select metabolite examples were illustrated.4 However, within the community of biotransformation, especially in the drug discovery setting, NMR plays a much smaller role than does MS. This is mostly attributed to two reasons. Firstly, while tandem MS allows the identification of metabolites present in complex in vitro incubations and in vivo biofluids, the sample for NMR has to be reasonably pure and largely free of endogenous contaminants to allow reliable and unambiguous assignment, typically requiring time-consuming metabolite scale-up and purification that ultimately restrict the frequent use of NMR. Secondly, although recent cryogenic probe technology significantly reduced thermal noise and increased instrument sensitivity and resolution by cooling the receiver electronics to near liquid helium temperature, NMR still is a relatively insensitive technique compared to MS. 1D NMR spectral acquisition typically requires a sample size of at least 0.5 μg, which may not even be sufficient for on-line LC-NMR. Although the expectation set for online LC-NMR since its emergence more than a decade ago had been high, to practicing biotransformation scientists, this technique has not lived up to its promise to simplify the structure determination of drug metabolites primarily due to limitations in chromatographic resolution and required sample size going to the flow probe cell.5 In order to overcome the common obstacles and impracticality of metabolite structure determination by NMR, we developed an improved analytical HPLC-based method for sample preparation, followed by structure determination by an NMR spectrometer equipped with a cryoprobe. Since the process is based on well established analytical scale chromatographic and NMR technologies, it allowed us to optimise each technique independently, provide sufficient NMR information on 0.5–10 μg of metabolite samples, and assign their structures on a regular basis. Herein, this paper is intended to describe the methodology for rapidly purifying microgram quantities of and biologically relevant metabolites for structure determinations using trazodone as a model compound.Experimental
Reagents and enzymes
NADPH, Tris-HCl, dibasic and monobasic potassium phosphate, ammonium acetate, and trazodone were of analytical grade and obtained from Sigma-Aldrich Inc. (St. Louis, MO, USA). HPLC grade acetonitrile, methanol and water were obtained from J. T. Baker (Phillipsburg, PA, USA). Pooled human liver microsomes or human recombinant CYP isoforms were purchased from BD Gentest (Woburn, MA, USA). The NMR solvent hexadeuterodimethyl sulfoxide (DMSO-d6, D 99.9%) was purchased from Cambridge Isotope Laboratories, Inc. (Andover, MA, USA).Liver microsome incubations of trazodone
Incubations (5 mL total volume) were performed in a shaking water bath at 37 °C. Trazodone (30 μM final concentration, 0.15 μmole) was pre-incubated for 5 min in Tris-HCl buffer (50 mM pH 7.4) containing microsomes (1 mg mL−1 protein content). NADPH (3 mM) and GSH (5 mM) were added to initiate reactions. Reactions were stopped at 90 min by addition of cold acetonitrile (3X volumes). Chemical stability of trazodone was assessed in incubations in which liver microsomes were not added. After centrifugation at 1500 g for 10 min, the extent of metabolite formation was monitored by HPLC-MS/MS analysis to be described subsequently while the remainder of the supernatants were evaporated overnight on a Centrivap and reconstituted in 0.8 mL of the mobile phase for metabolite purification.HPLC and metabolite purification
HPLC system consisted of Thermo Accela quaternary pump, autosampler and photodiode array detector (ThermoFisher Scientific, San Jose, CA, USA). Chromatography was carried out on a Waters Sunfire C-18 column (3 mm × 150 mm, 3 μm) with a binary mixture of 10 mM ammonium acetate adjusted to pH 5.0 with formic acid and containing 5% acetonitrile (solvent A) and acetonitrile (solvent B). The mobile phase initially consisted of A/B (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10), it was then linearly programmed to A/B (10:90) over 25 min, held for 5 min and then programmed to A/B (90:10) over 1 min. The column was equilibrated at A/B (90:10) for 5 min before making the next injection. A flow rate of 0.4 ml min−1 was used for all analyses. For metabolite purifications, fractions from 12–22 min during the LC run were collected into a deep well microtiter plate containing 0.7 mL glass inserts at 0.125 min per well. Multiple injections (60 μL each) and chromatographies (up to 10) were repeated under the same conditions in an automated fashion, and eluent from the column were collected into the same plate. The solvent in collected metabolite wells was evaporated overnight on a Centrivap prior to dissolution in deuterated solvent for NMR.
:10), it was then linearly programmed to A/B (10:90) over 25 min, held for 5 min and then programmed to A/B (90:10) over 1 min. The column was equilibrated at A/B (90:10) for 5 min before making the next injection. A flow rate of 0.4 ml min−1 was used for all analyses. For metabolite purifications, fractions from 12–22 min during the LC run were collected into a deep well microtiter plate containing 0.7 mL glass inserts at 0.125 min per well. Multiple injections (60 μL each) and chromatographies (up to 10) were repeated under the same conditions in an automated fashion, and eluent from the column were collected into the same plate. The solvent in collected metabolite wells was evaporated overnight on a Centrivap prior to dissolution in deuterated solvent for NMR.
HPLC-MS/MS
HPLC-MS/MS was conducted on a Thermo Orbitrap Discovery using electrospray ionisation. The eluent from the HPLC column was routed into the flow cell of a Thermo Surveyor photodiode array detector, then into the ion source of the mass spectrometer. The delay in response between the two detectors was about 0.2 min. The electrospray interface was operated at 3 KV, sheath pressure at 80 PSI, capillary temperature at 275 °C and the mass spectrometer was operated in the positive mode. High resolution full scan MS measurements were obtained on the Orbitrap at 30,000 resolution (at 500 Da) with daily external calibrations using a mixture of ultramark, MRFA and caffeine as specified by the vendor. MSn studies were performed using the linear ion trap (LTQ) with an isolation width of 3, normalised collision energy of 30V, an activation Q of 0.25 and an activation time of 30 msec. MSn fragments were passed into the Orbitrap for high resolution mass measurements.NMR
Each metabolite sample, collected and dried in the 0.7 mL glass insert from HPLC runs, was dissolved in 50 μL DMSO-d6 and transferred to a 1.7 mm NMR tube. The use of 1.7 mm tube for metabolite analysis was driven by the reduction of interference noise from NMR solvent and residual water, although the mass sensitivity gain from changing 3 mm to 1.7 mm tube in a 5 mm probe was insignificant since the rf coil was further away from the sample.6,7 All NMR data were acquired on a Bruker 500 MHz spectrometer equipped with a 5 mm TCI cryoprobe. Metabolite quantity was calculated by comparing the integral of proton signals of metabolites to the integral of 13C satellite peaks of DMSO-d6, namely the QSQC method described by Molinski and co-workers.8 Briefly, a quantification curve was established by using 2.0, 1.0, 0.5, 0.2 and 0.1 mM of trazodone as an external standard. Using the equation ACH/Asat = c× m., where ACH/Asat is the integral ratio of an aromatic proton peak of trazodone and the 13C satellite peaks of DMSO-d6, m is number of moles of trazodone, the slope (c) was determined as 0.34 nmol−1 (R = 0.98927) and subsequently used for the calculation of metabolite quantity. The quantity of metabolites, m, was obtained from the same equation where ACH/Asat was the integral ratio of the aromatic proton peak of the metabolite and the 13C satellite peaks of DMSO-d6. To assist in the interpretation of NMR spectra, ACD 1H-NMR Predictor (Advanced Chemistry Development, Toronto, ON) was used.Results and discussion
Trazodone is a second generation antidepressant and anxiolytic. It inhibits the re-uptake of serotonin, but possesses a lower affinity for the serotonin transporter (SERT) than selective serotonin reuptake inhibitors (SSRI), such as fluoxetine. Trazodone's anxiolytic and antidepressant effects are likely due to its antagonism at the 5-HT2A and 5-HT2C receptors. It is clinically used for the treatment of major depression, often in conjunction with fluoxetine or other SSRIs, or to control sleep disturbance symptoms when using SSRIs and norepinephrine reuptake inhibitors (SNRIs) because of its sedating side effects.10Trazodone is extensively metabolised in humans via hydroxylation, N-dealkylation, and N-oxidation pathways.11–14 Early biotransformation studies of trazodone led to the identification of several major metabolites in the human and animal excreta, including a triazolopyridinone dihydrodiol metabolite (Met5), a chlorophenyl hydroxyl metabolite (Met4) and its glucuronide, whereas in human plasma, 1-(3′-chlorophenyl)piperazine or m-CPP (Met3), was a major circulating metabolite. Several recent investigations focused on the metabolic activation of trazodone to electrophilic quinine-imine and epoxide intermediates at the chlorophenyl and triazolopyridinone rings, respectively.15,16 A number of glutathione (GSH) conjugates were identified from NADPH-supplemented microsomes in the presence of GSH. However, except Met3–5, most metabolite structures were assigned tentatively based on the combination of LC-MS/MS spectra and proposed bioactivation reaction mechanism. Without NMR evidence or direct comparison with synthesised metabolite standards, the regiochemistry of these metabolites remained unclear. The pattern of multi-site metabolism and formation of both oxidative and conjugative metabolites make trazodone a good model compound to apply our methodology to microgram scale metabolites for rapid structure determination using NMR.
Metabolite generation and NMR sample preparation
The generation of oxidative metabolites and GSH conjugates of trazodone in NADPH-supplemented microsomes in the presence of GSH was achieved by a single 5 mL incubation using 30 μM of trazodone. The incubation conditions, post incubation work-up, and sample preparation by HPLC closely resembled a typical biotransformation experiment. The HPLC-MS/MS system used mild or low concentration of mobile phase modifier (10 mM ammonium acetate). Other HPLC conditions for purification of metabolite were also very similar to typical metabolite identification analysis except that multiple (5–10) injections and separations of metabolites were made overnight in an automated fashion to reduce column overloading. Fractions from HPLC runs were collected repeatedly into a deep well microtiter plate containing 0.7 mL glass inserts for drying in Centrivap while heat and high intensity light were avoided. Glass inserts were used for metabolite collection and transfer to a small volume (50 μL) of deuterated solvents for NMR since they had significant advantage in minimising sample contamination from phthalates which were frequently encountered in commercially available microtiter plates and caused interference in NMR spectra. If metabolites or conjugates are anticipated to be unstable due to acidic or basic HPLC mobile phase modifier during sample work-up, addition of buffer solution, such as 1 M ammonium acetate (pH 6.5, 100 μL) into the glass inserts prior to fraction collection was found to greatly reduce the sample decomposition. The established HPLC purification process reliably yielded 0.5–10 μg of metabolites8 of trazodone and other drug candidates in our laboratories, which were sufficient for most 1D 1H-NMR and some 2D-NMR with satisfactory signal-to-noise ratio. Most structure determinations were achieved by interpreting diagnostic 1D 1H-NMR spectral information associated with the site of metabolism, such as chemical shifts, 1H-1H coupling patterns, and integrations. For proper spectral comparison among metabolites while avoiding data inconsistency caused by extraneous pH, solvent, and other HPLC related effects, the NMR spectra of metabolites were directly compared to the spectrum of trazodone which was prepared from the same HPLC experiment.Seven fractions containing a total of eight metabolites and GSH conjugates of trazodone in human liver microsomes were obtained from the HPLC runs, and were subjected to NMR data acquisition using a 5 mm cryoprobe (1.7 mm tube). Both LC-MS/MS and NMR spectra indicated that a pair of metabolites, Met1a/Met1b, were not fully separable on HPLC and collected into a single fraction.
Metabolite characterisation
The accurate mass spectrum (MS), MS/MS and NMR spectral data characteristic to the metabolites are shown in Table 1, and expanded 1H-NMR spectra of aromatic region in Fig. 1. For NMR signal assignment purpose, the numbering of structural elements is intended for illustration only and does not comply with IUPAC rules.| Met# | HPLC Rt/min | Observed MH+ (m/z) | Mass error Δppma | Empirical Formula | Estimated quantity (μmole)8 | Key MS/MS fragment ions (m/z) | Key NMR information | Combined data interpretation on metabolite structure | Regiochemistry determination by NMR |
|---|---|---|---|---|---|---|---|---|---|
| a The mass error Δppm = 1 × 106 (observed mass of MH+ - theoretical mass of MH+)/theoretical mass of MH+. b Combined weight of predominant Met1a and minor Met1b. c Under LC-MS conditions, trazodone had an HPLC retention time (Rt) of 22.5 min. | |||||||||
| Met1a | 11.7 | 262.1661 | 0.5 | C13H19N5O | 0.007b | 197,176,165, 148 | H5, H6, H7 and H8 resonances of the triazolopyridinone moiety | N-dealkylation of chlorophenyl ring | Yes |
| Met1b | 11.7 | 693.2203 | 1.9 | C29H38O8N8ClS | 675,564,420, 388,176 | GSH conjugate on chlorophenyl | No | ||
| Met2 | 16.5 | 695.2358 | 2.1 | C29H40O8N8ClS | 0.009 | 677,659,566, 422,237 | H5, H6, H7 and H8 on partially reduced triazolopyridinone moiety from 1D NMR and 1H-1H COSY | 7-OH, 8-GSH conjugate on partially reduced triazolopyridinone | Tentative |
| Met3 | 17.2 | 197.0850 | 5.0 | C10H14ClN2 | 0.048 | 154 | Intact chlorophenyl ring, but no proton signals due to triazolopyridinone | 1-(3′-chlorophenyl) piperazine or m-CPP | Yes |
| Met4 | 18.2 | 388.1531 | 0.9 | C19H22O2N5Cl | 0.026 | 350,251,176, 148,133 | H18, H21, H22 on chlorophenyl ring, H18/H15 and H22/H15 NOEs in ROESY | para-hydroxyl (20-OH) on chlorophenyl | Yes |
| Met5 | 20.2 | 406.1634 | 1.6 | C19H24O3N5Cl | 0.003 | 386,235,210, 192,182,164 | H5, H6, H7 and H8 assigned to partially reduced triazolopyridinone from 1D NMR and 1H-1H COSY | 7-OH, 8-OH dihydrodiol on triazolopyridinone | Yes |
| Met6 | 21.3 | 406.1637 | 0.7 | C19H24O3N5Cl | 0.0002 | 386,210,192, 182 | 1H-NMR not interpretable | Suspected diastereomer of Met5 | No |
| Met7 | 21.6 | 388.1530 | 1.2 | C19H22O2N5Cl | 0.013 | 253,192,176, 148,133 | 1H-NMR similar to Met 4, but H18, H20 and H21 on chlorophenyl ring in a different ABC spin system from that of Met4. | 22-OH on chlorophenyl | Tentative |
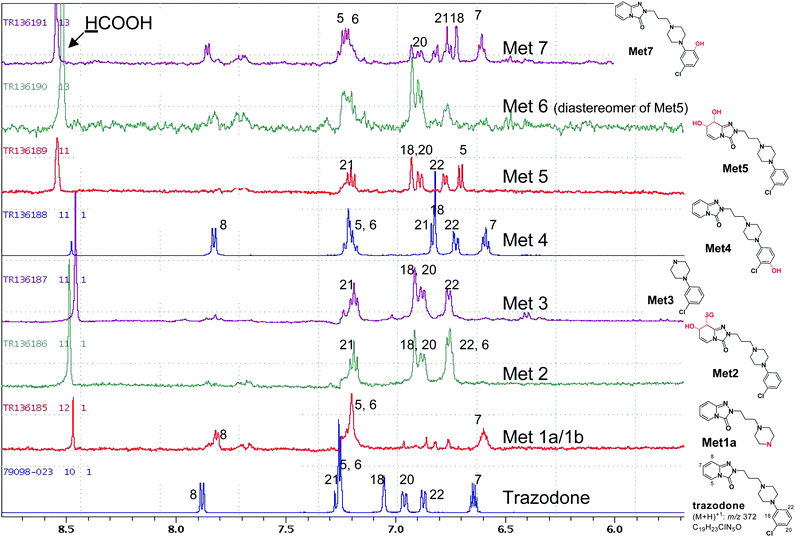 | ||
| Fig. 1 Expanded 1H-NMR spectra of aromatic region of trazodone metabolites in human liver microsomes. 1H-NMR experiments were performed by using the wet pulse sequence for water suppression.9 Typical NMR parameters include 256 numbers of scan; 1 Hz line broadening; 1 s delay time; and 1.6 s acquisition time. | ||
The fraction (1.8 μg/0.007 μmole) containing Met1a and Met1b showed the 1D 1H-NMR spectrum originated almost completely from Met1a, which exhibited a protonated molecular ion (MH+) at m/z 262.1661 and an empirical molecular formula of C13H19N5O from accurate MS, indicating the loss of chlorophenyl moiety due to metabolism. The loss of the moiety was also evident in the 1H-NMR (Fig. 1) where only four aromatic proton resonances of the triazolopyridinone moiety, H5 (7.20 ppm), H6 (7.2 ppm), H7 (6.6 ppm) and H8 (7.85 ppm), were observed. Interestingly, Met1a has never been reported as a trazodone metabolite prior to the present study.
Met1b was a minor metabolite in the fraction. Its accurate MS/MS showed a molecular ion (MH+) at m/z 693.2203 and empirical molecular formula of C29H38O8N8ClS, indicative of a GSH conjugate in conjunction with hydroxylation. In addition to fragment ions at m/z 564 (−129 Da, -glutamyl) and m/z 420 (−273 Da, -glutathionyl lacking sulfur) diagnostic for an aromatic (sp2) carbon-linked GSH conjugate, the product ion at m/z 176 corresponded to an intact N-propyl-triazolopyridinone piece, suggesting that hydroxylation and subsequent GSH conjugation had occurred on the 3-chlorophenyl ring system. Met1b appeared to be identical to the “conjugate 6” reported by Kalgutkar and co-workers.15
However, the regiochemistry of GSH conjugation and hydroxylation remained undefined due to insufficient quantity of sample.
Met2 proved to be another GSH conjugate of trazodone as indicated by its molecular ion (MH+) at m/z 695.2358 and empirical molecular formula of C29H30O8N8ClS. Its MS/MS product ion spectra, including the diagnostic fragment ion at m/z 237 representing an intact 3-chlorophenyl-N-propylpiperazine moiety, led to the assignment of Met2 as a “dihydrodiol” type GSH conjugate at the triazolopyridinone motif, which seemed to be similar to the “conjugate 3” reported by Kalgutkar et al.15 but without regiochemistry characterisation. The 1H-NMR spectrum of Met2 exhibited resonances of two vinyl protons H5 (6.75 ppm, d) and H6 (5.59 ppm, m) that significantly shifted upfield compared to those of trazodone (Fig. 1, Fig. 2) whereas those of H7 and H8 protons also shifted upfield due to loss of the double bond and were masked by the intense water and solvent peaks in the 1D 1H-NMR spectrum. A subsequent 1H-1H COSY spectrum revealed H7 (4.9 ppm) and H8 (5.38 ppm), and further extended the H5/H6 spin system to H6/H7 and H7/H8 (Fig. 2). Using ACD software for the prediction of 1H chemical shifts based on NMR data of similar sub-structures, the observed chemical shifts of H5, H6, H7 and H8 were more consistent with the predicted values of a 7-hydroxyl, 8-glutathionyl substitution rather than a 7-glutathionyl, 8-hydroxyl substitution (Table 2). Met2 is thus proposed to have 7-hydroxyl and 8-glutathionyl regiochemistry on the partially reduced triazolopyridinone moiety.
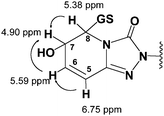 | ||
| Fig. 2 Key correlations observed in the 1H,1H-COSY spectrum of Met2. | ||
The major metabolite Met3 (9.4 μg/0.048 μmole) demonstrated a molecular ion (MH+) at m/z 197.0850 and empirical molecular formula of C10H14ClN2, identical with those reported for 1-(3′-chlorophenyl)piperazine or m-CPP, an N-dealkylation product present in human plasma. The assignment of Met3 to m-CPP was also supported by its 1H-NMR spectrum where only proton signals from an intact chlorophenyl ring were observed in the aromatic region (Fig. 1).
Met4 was among the most abundant metabolite (9.9 μg/0.026 μmole) obtained from the human liver microsomal incubation of trazodone. Its molecular ion (MH+, m/z 388.1531), empirical molecular formula (C19H22O2N5Cl), and MS/MS data (Table 1) were consistent with a mono-hydroxylated metabolite on the chlorophenyl ring. The 1H-NMR spectrum of Met4 exhibited a number of key features; a recognisable ABC coupling pattern among three remaining protons on the chlorophenyl ring, and marked upfield shifts of H21 (6.83 ppm) and H18 (6.81 ppm) from corresponding H21 (7.20 ppm) and H18 (7.02 ppm) in trazodone (Fig. 1), that suggested that Met4 is either a 20-hydroxyl or a 22-hydroxyl metabolite. The concrete regiochemistry evidence was obtained by 2D-ROESY (rotating frame Overhauser enhancement spectroscopy) spectrum, which clearly indicated through space interactions (NOEs) between H18 and H15, and H22 and H15 (Fig. 3), leading to the final assignment of 20-hydroxyl or para-hydroxy trazodone structure for Met4, which was previously detected in human urine,11,13 rat excreta,12 and liver microsomal incubations.15
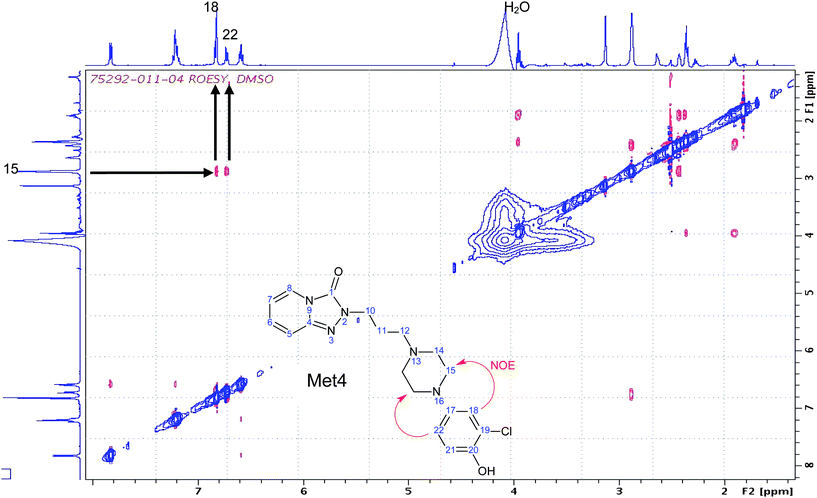 | ||
| Fig. 3 Diagnostic region of ROESY of Met4. ROESY spectrum of ∼26 nmole of Met4 dissolved in 50 μL of DMSO-d6 was acquired as 2048 × 128 matrix, with 64 scans per increment. A 2D ROESY with cw spin lock for mixing pulse sequence were used for this experiment.17 The data was processed to 2048 × 2048 points by zero-filling the second frequency domain. | ||
Both Met5 and Met6 showed the same molecular ion (MH+, m/z 406) and empirical molecular formula (C19H24O3N5Cl), suggesting two dihydrodiol metabolites. The observed product ions at m/z 210, 192, 182, 164 (Table 1) further suggested the location of dihydrodiol at the triazolopyridinone ring.15 However, only Met5 exhibited interpretable 1H-NMR spectrum (Fig. 1). Four triazolopyridinone proton signals of Met5, H5 (6.60 ppm), H6 (5.51 ppm), H7 (4.15 ppm), and H8 (4.38 ppm), were found to belong to one spin system from the 1H, 1H-COSY spectrum (data not shown), and were also significantly shifted upfield compared to corresponding protons in trazodone. Furthermore, the lack of the characteristic trazodone H8 signal near 7.88 ppm in Met5 implied cytochrome P450 mediated oxidation at C7 and C8 positions. The combined evidence strongly supported a 7-, 8-dihydrodiol rather than a 5-, 6-dihydrodiol for Met5 although the relative stereochemistry of C7 vs. C8 could not be determined in this study. The assigned regiochemistry is also consistent with that reported for a major trazodone metabolite in human urine.11,13,14
Met7 showed identical molecular ion (MH+, m/z 388.1531), empirical molecular formula (C19H22O2N5Cl), and product ion spectra (Table 1) to Met4. Met7 is thus a mono-hydroxylated metabolite on the chlorophenyl ring and a regioisomer of Met4. The 1H-NMR spectrum of Met7 was similar to that of Met 4, except that the ABC spin system of the chlorophenyl protons in Met7, comprising H18 (6.68 ppm), H20 (6.79 ppm) and H21 (6.73 ppm), shifted noticeable from that of Met4. Consequently, Met7 was proposed as the 22-hydroxyl metabolite of trazodone (Fig. 1).
In summary and as shown in Fig. 4, the structure and regiochemistry of four metabolites, Met1a, Met3, Met4 and Met5, were unambiguously characterised using 1H-NMR, 1H, 1H-COSY, and ROESY techniques on 1–10 μg of sample in combination with complimentary LC-MS data. Regiochemistry assignments for another two metabolites (Met2 and Met7) were proposed based on observed chemical shifts and similarities to other metabolites. Two minor metabolites, Met1b and Met6, were not structurally defined beyond the gross structure assigned from MS/MS data primarily due to insufficient quantity.
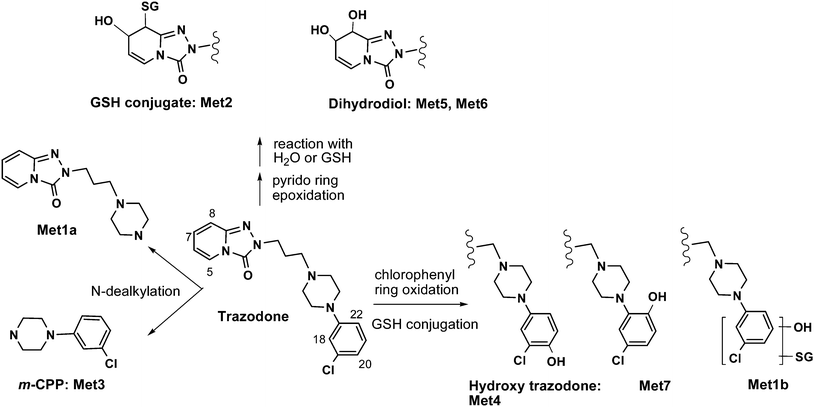 | ||
| Fig. 4 Trazodone metabolites in human liver microsomes identified by microgram scale NMR. | ||
Among the reported trazodone metabolites, only the regiochemistry of Met4 and Met5 was previously characterised.11,13,14,15 Thus, the present study provided the first regiochemical evidence for Met2 and Met7 from NMR. In addition, the structure of Met1a has not been published previously and is thus established here for the first time.
It is important to point out that all the NMR data acquisitions and structure elucidations were achieved on microgram scale metabolites originated from a single 5 mL microsomal incubation of trazodone (30 μM) and completion time for the entire workflow was within a few consecutive days. At similar substrate concentration (30 μM) and incubation size (1–5 mL), the general criteria for a successful NMR experiment are 10% or more metabolite formation, where the theoretical yield of a metabolite with molecular weight of 500 would be in the 1.5–7.5 μg range.
As a comparison, we also evaluated a microflow probe NMR (10 μL active volume, Protasis Probe) with a sample isolated from HPLC. It required typically a larger sample size (5 μg or more) for 1H-NMR spectrum due to dilution effect from the push solvent and a less sensitive receiver operating at the room temperature. An overnight ROESY data acquisition on 5 μg trazodone sample failed to provide interpretable dataset. The shortcoming due to dilution effect of the microflow probe may be improved with segmented flow technology18 when it becomes more accessible. Similar to LC-NMR, the microflow method suffered from greater difficulty in sample recovery.
Experience suggests that the most significant challenge in online LC-NMR, either in a stop-flow or loop-storage mode, remains the design of an appropriate chromatographic system to separate interfering substances while capturing the eluting peak of interest into the flow probe cell. Optimally, the peak volume should be matched to the flow cell volume, which often requires extremely fine control of system timings. In addition, the typical flow cell volume (∼120 μL) is at least 2-times larger than that of 1.7 mm NMR tube, leading to reduced sensitivity due to sample dilution. Another disadvantage of stop-flow mode occurs if multiple metabolites are analysed. The metabolite peaks still remaining on the HPLC column may deteriorate during the NMR data acquisition time. LC-solid phase extraction (SPE)-NMR addresses some limitations of LC-NMR since SPE provides a simpler approach to collect and concentrate individual HPLC peaks prior to washing off the analyte directly into a conventional NMR tube.19,20 However, not all metabolites, particularly water-soluble GSH conjugates and glucuronides, can be readily trapped onto an SPE cartridge.
We estimated metabolite quantities using the recently published QSQC method by comparing the integral of proton signal of metabolites to the integral of 13C satellite peaks of DMSO-d6.8 It should be pointed out that although this method provides useful quantity information for most metabolites, there are complications when applied to microgram level of metabolites. Unlike neat chemicals that can be accurately weighed and serially diluted in a non-aqueous NMR solvent, the metabolites were prepared from reversed phase HPLC fractions. The wetness of samples, particularly those early eluted metabolite peaks, is often significant and detrimental to quality 1H-NMR spectra. Many NMR factors, such as S/N ratio, resolution and sharpness of the proton signal used for calculation, degree of water suppression, duration of the relaxation delay, imperfect peak shape due to potential intermediate exchange can inevitably introduce error for metabolite quantification. Nevertheless, in the drug discovery environment, the estimated metabolite quantity using this method offers valuable information in the rank-order of metabolites without synthetic standards.
Conclusion
In the present study we successfully developed a robust methodology for rapid structure determination of microgram scale drug metabolites using cryoprobe NMR spectroscopy. Interpretable 1H-NMR spectra of metabolites can be obtained in a time- and cost–effective manner starting from routine scale incubation (1–5 mL) with a parent compound (10–30 μM) in microsomes, hepatocytes, or recombinant drug metabolising enzymes, typically cytochrome P450s and UDP-glucuronosyltransferases. The utilisation of a high field NMR equipped with a 5.0 mm cryoprobe (1.7 mm tube) extended the limit of detection and resulted in adequate 1H-NMR with ∼1 μg or more sample after isolation by analytical HPLC. 2D NMR experiments may be needed for structure elucidation of some metabolites and sample requirements would increase to ∼5–10 μg depending on the complexity of the structure, sharpness of NMR signals and the need of unmasking solvent interference. Since our off-line methodology is based on well established chromatographic and NMR technologies, it allowed us to optimise each technique independently, and made it practical to obtain sufficient NMR information fast and on a regular basis without having to develop preparative or NMR-compatible HPLC separations. A large number of examples, including that of trazodone, from our laboratories have proven that the microgram-level NMR method avoids time consuming preparative-scale metabolite generation and purification and circumvents technical complications associated with online LC-NMR. Most importantly, the turnaround time of the metabolite structure determination workflow becomes in sync with the cycle time during which medicinal chemists modify metabolic softspots while performing other iterative lead optimisation activities, demonstrating a real impact on the drug-discovery process.References
- Y. Z. Shu, B. M. Johnson and T. J. Yang, AAPS J., 2008, 10, 178–192 Search PubMed.
- S. Ma, S. K. Chowdhury and K. B. Alton, Curr. Drug Metab., 2006, 7, 503–523 Search PubMed.
- A. P. Watt, R. J. Mortishire-Smith, U. Gerhard and S. R. Thomas, Curr. Opin. Drug Discovery Dev., 2003, 6, 57–65 CAS.
- X. Huang, R. Powers, A. Tymiak, R. Espina and V. Roongta, in Drug Metabolism in Drug Design and Development ed. D. Zhang, M. Zhu and W. G. Humphreys, John Wiley & Sons, Inc., New York, 2007, pp. 369–409 Search PubMed.
- Y. Chen, M. Monshouwer and W. L. Fitch, Pharm. Res., 2007, 24, 248–257 CrossRef CAS.
- G. E. Martin and C. E. Hadden, J. Nat. Prod., 2000, 63, 543–585 CrossRef CAS.
- T. M. de Swiet, J. Magn. Reson., 2005, 174, 331–334 CrossRef CAS.
- D. S. Dalisay and T. F. Molinski, J. Nat. Prod., 2009, 72, 739–744 CrossRef CAS.
- R. J. Ogg, P. B. Kingsley and J. S. Taylor, J. Magn. Reson., Ser. B, 1994, 104, 1–10 CrossRef CAS.
- M. Haria, A. Fitton and D. McTavish, Drugs Aging, 1994, 4, 331–355 CrossRef CAS.
- L. Baiocchi, A. Frigerio, M. Giannangeli and G. Palazzo, Arzneimittelforschung, 1974, 24, 1699–1706 CAS.
- C. Yamato, T. Takahashi and T. Fujita, Xenobiotica, 1974, 4, 313–326 CrossRef CAS.
- R. Jauch, Z. Kopitar, A. Prox and A. Zimmer, Arzneimittelforschung, 1976, 26, 2084–2089 CAS.
- S. Fujiwara, K. Noumi, T. Kawashima and N. Awata, Yakuri to Chiryo, 1989, 17, 1365–1382 Search PubMed.
- A. S. Kalgutkar, K. R. Henne, M. E. Lame, A. D. Vaz, C. Collin, J. R. Soglia, S. X. Zhao and C. E. Hop, Chem.-Biol. Interact., 2005, 155, 10–20 CrossRef CAS.
- B. Wen, L. Ma, A. D. Rodrigues and M. Zhu, Drug Metab. Dispos., 2008, 36, 841–850 CrossRef CAS.
- D. Bax and A, D.G., J. Magn. Reson., 1985, 63, 207–213 CAS.
- R. A. Kautz, W. K. Goetzinger and B. L. Karger, J. Comb. Chem., 2005, 7, 14–20 CrossRef CAS.
- M. Godejohann, L. H. Tseng, U. Braumann, J. Fuchser and M. Spraul, J. Chromatogr. A, 2004, 1058, 191–196 CrossRef CAS.
- M. Sandvoss, B. Bardsley, T. L. Beck, E. Lee-Smith, S. E. North, P. J. Moore, A. J. Edwards and R. J. Smith, Magn. Reson. Chem., 2005, 43, 762–770 CrossRef CAS.
Footnote |
| † Y.-Z. Shu wishes to dedicate this article to Professor Masao Hattori on the occasion of his retirement from Toyama University, Japan |
| This journal is © The Royal Society of Chemistry 2010 |