Using a portable device based on a screen-printed sensor modified with a molecularly imprinted polymer for the determination of the insecticide fenitrothion in forest samples
Cristina
Pellicer
,
Alberto
Gomez-Caballero
,
Nora
Unceta
,
M. Aranzazu
Goicolea
and
Ramón J.
Barrio
*
Department of Analytical Chemistry, Faculty of Pharmacy, University of the Basque Country, 01006, Vitoria-Gasteiz, Spain
First published on 8th July 2010
Abstract
A molecularly imprinted polymer is presented as a screen printed electrode (SPE) coating for determining the presence of fenitrothion in environmental samples for selective and in situ analysis. The polymeric film was obtained by electropolymerization of Ni(II)-phtalocyanine in presence of the template molecule, through the use of cyclic voltammetry. The electropolymerization process as well as the measurement of the fenitrothion by square wave voltammetry (SWV) were studied and optimized. The response of the modified SPE was linearly proportional to concentration of fenitrothion over the range of 3 × 10−6 to 10−4 mol L−1, with a LOD of 8 × 10−7 M and good stability and reproducibility (RSD < 14%). The selectivity of the sensor was tested by measuring fenitrothion in the presence of some possible interferent compounds such as metamitron, fenitrothion-oxon and methyl-parathion obtaining satisfactory results. The device used favours the portability of the equipment which can be used to carry out “in situ” analysis of fenitrothion in forest matrices.
Introduction
One of the main challenges facing the analytical chemist is to develop methods that respond to the growing need to perform rapid ‘in situ’ analyses to minimize the effects of sampling and transportation and to avoid all the pre-treatments. These methods must be sensitive and accurate, and able to determine substances in ‘real-life’ samples. In recent years, many of the methods developed with this end in sight have been based on the use of electrochemical sensors due to their high sensitivity and selectivity, portable field-based size and low-cost.1,2Screen printing is a technique that permits the miniaturization of sensors, making it possible to integrate the reference and working electrodes in the same chip. Furthermore, thanks to the low cost and reduced size of the device manufactured by screen printing, the screen printed electrodes (SPEs) are easily associated to a simple, economic and portable electrochemical instrumentation, which makes them ideal for on-site environmental analysis in real time and with null or low generation of residues.3,4 The great versatility presented by the SPEs lies in the wide range of ways in which the electrodes may be modified. The composition of the printing inks may be altered by the addition of very different substances such as metals, enzymes, polymers or complexing agents.5 On the other hand, there also exists the possibility of modifying the manufactured electrodes by means of depositing various substances on the surface of the electrodes such as metal films,6 nanoparticles7–10 polymers,11,12 or enzymes.13,14
Molecular imprinting is a consolidated way to create artificial recognition sites in synthetic materials, improving the selectivity of the analytical determinations. This technique lies in synthesizing substrate-selective recognition sites in a polymeric matrix using a molecular template during the polymer formation.15 After the removal of the entrapped template from the polymeric matrix, molecular prints complementary to the target compound still remain. In subsequent experiments these imprinted sites, will be able to selectively recognize the target analyte. Electrochemical sensing with electrodes modified with MIP films have been developed for the determination of different substances.16–19 MIPs based on screen-printed sensors, for the detection of pesticides and other substances, where the formation of the polymer is made using the traditional method of radical polymerization, have been previously developed20–23
In recent years, interest has been increasing in electropolymerization as a new strategy with which to carry out MIP generation.24 Electrosynthesis of MIP based sensors, allows the generation of a rigid and compact polymeric mesh and the polymerization process can be controlled obtaining a uniform and thick surface.25,26
The aim of this study was to modify the surface of screen-printed carbon-based electrodes (SPCEs) with an electrogenerated molecularly imprinted polymer, allowing greater reproducibility in obtaining the electrode coating. The SPCE was modified by electrodeposition of poly Ni(II)-phthalocyanine in the presence of fenitrothion which acted as a template molecule. The chip manufactured in this way enables the selective determination of the insecticide fenitrothion in environmental samples. The stability and simplicity of the device favours the portability of the equipment which can be used in field studies covering large forest areas in order to establish dosages of pesticide linked to geographic coordinates provided by a GPS system.
Experimental
Chemicals and reagents
The pesticides fenitrothion, fenitrothion-oxon, methyl parathion and metamitron were purchased from Dr Ehrenstorfer GmbH (Augsburg, Germany). Stock solutions of these pesticides were prepared in reagent grade methanol (Scharlab, Barcelona, Spain) and stored at −40 °C. Ni-phtalocyanine was obtained from Sigma-Aldrich (St. Louis, USA) and employed as received.Folithion 50 EC (Bayer CropScince, Alcacer, Spain) with 50% fenitrothion p/v was used in real sample studies.
All the electroanalytical measurements were performed at room temperature using Britton-Robinson buffer as a supporting electrolyte. The buffer was obtained by mixing appropriate amounts of orthophosphoric acid 85%, acetic acid 96% and boric acid obtained from Merk (Darmstad, Germany). NH4OH/NH4Cl buffer was used to extract the template molecule from the polymer, and was obtained by mixing NH4OH 25% and NH4Cl, both from Panreac (Barcelona, Spain). All other reagents were analytical reagent grade. All solutions were prepared using doubly-deionized water Milli-Ro and a Milli-Q water purification system (Millipore, Bedford, MA, USA).
Apparatus
In order to enhance the portability of the device, a 175 g potentiostat μStat 200 (Drop Sens S.L. Oviedo, Spain) was used with the following dimensions L8.5 × W5.4 × H2.3 cm, This was connected via a USB connection to laptop computer installed with the Measurement Software DropView (DropSens). The entire set could easily be transported in a light and compact briefcaseScreen printed electrodes (Drop Sens, DRP-110) were constituted by a ceramic substrate (L33 × W10 × H0.5 mm) that incorporates the electrochemical cell, consisting of a working electrode (carbon, 4 mm diameter), a counter electrode (carbon) and a reference electrode (silver) (Fig. 1)
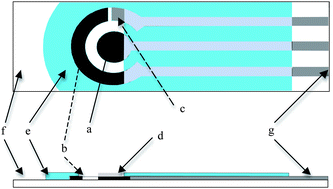 | ||
| Fig. 1 (a) Carbon Working Electrode, (b) Reference electrode, (c) Counter electrode, (d) MIP layer, (e) insulation, (f) ceramic substrate, (g) silver leads. | ||
Development of the MIP based sensor
Pretreatment is a procedure aimed not only at increasing the sensitivity of the sensor, but also at obtaining a stable baseline for long term experiments. For this reason, initially, SPCEs were pretreated by sweeping the potential between 0.0 and −1.0 V in 0.5 mol L−1 H2SO4 at 0.1 V s−1 until a reproducible voltammogram was observed.27To carry out the surface modification of the sensor, Ni(II)-phtalocyanine was chosen as the functional monomer. It has been used previously as an electrode surface modifier28 due to the excellent stability and the electrochemical properties that the resulting conducting polymer has. The electrosynthesis of the MIP-sensor was performed by cyclic voltammetry in a potential range between −0.1 and 0.60 V at 0.05 V s−1 for 75 cyclic scans. A drop of 40 μL of a solution of 5 × 10−5 mol L−1 Ni(II)-phthalocyanine in 0.05 mol L−1 NaOH containing 5 × 10−5 M of fenitrothion was used as a polymerization medium.
Following the film deposition, the fenitrothion trapped in the polymer matrix was extracted by immersion in a stirring solution of NH4OH/NH4Cl 0.1 M buffer at 50 °C for 15 min. A non-imprinted sensor (NIP), employed as a control sensor, was synthesized exactly in the same way but without the addition of the template molecule into the polymerization mixture. After the extraction of the template, the sensor was ready to be employed in the rebinding step.
Electrochemical measurements: Rebinding of the analyte
Previous to the electroanalytical measurement, an incubation stage is necessary. To this end 40 μL of the sample at an ionic strength of 1 M of NaCl, was deposited on the surface of the SPCE-MIP for a period of time, in order to achieve the union of the fenitrothion in the complementary gaps within its structure present in the polymeric mesh. Following the incubation period, the SPCE-MIP was cleaned with water, to eliminate the fenitrotion faintly adsorbed onto the electrode surface.All electroanalytical determinations were made at room temperature (20 ± 2 °C), on a drop (40 μL) of Britton-Robinson (BR) buffer at pH 1.8. The electrochemical measurements were carried out using square wave voltammetry (SWV) in the potential range between 0 and −1.0 V, at a frequency of 20 Hz, pulse amplitude of 110 mV and a step potential of 9 mV.
Studies with real samples: One drop analysis
To simulate field studies a spray dispenser (30 mL, AZLON, England) of fenitrothion was used. 10 g of black poplar leaves were collected and spread over laboratory table and the pesticide was applied, ensuring that the entire 5 mL of the solution (40 mg L−1 or 80 mg L−1 depending of the dosage) in the spray container was used. Following application, the samples were left to dry for at least 2 h before performing the extraction process. To this 0.5 mL of 1 M NaCl solution was added on the underside of poplar leaves and then it was left to rest for 10 min. 40 μL of this solution were deposited on the surface of the SPCE- MIP for incubation. After 10 min the electrode surface was washed with water, and the voltammetric measurement was made in 40 μL of BR buffer.Results and discussion
The parameters affecting the electrochemical reduction process of the nitro group of fenitothion in SPCE using SWV, were optimized. It is well know that the electrochemical reduction of the nitro group lead to a hydroxylamine group that can then be reversibly oxidized at a more anodic potential.29 Conditions of potential range between 0 and −1.0 V, at a frequency of 20 Hz, pulse amplitude of 110 mV and a step potential of 9 mV were chosen. The influence of pH of the supporting electrolyte in the SW response of a 10−4 M solution of fenitrothion was studied in BR buffer, in the 1–11 range. The electrochemical signal (Ip) decreased with increasing pH, for this a pH = 1.8 was fixed for the rest of studies. As it is was expected, Ep turns more cathodic with increasing pH, which indicates a contribution of H+ in the reduction electrochemical process.30 In these conditions, a well defined cathodic wave around −0.8 V was observed, which will be used in subsequent assays for the quantification of fenitrothion.Electropolymerization of Ni(II)-phtalocyanine on SPCEs
The electropolymerization of the monomer to obtain the Ni(II)-phtalocyanine film was carried out by cycling the potential under the previously mentioned procedure. Fig. 2a shows the growth patterns for Ni-phthalocyanine in continuous scan cyclic voltammetry. As can be seen, peaks observed at 0.28 and 0.35 V, correspond to the Ni(II)/Ni(III) redox couple. Deposition of polymeric Ni(II)-phtalocyanine onto the electrode surface during each successive scan can be confirmed by the increasing of anodic and cathodic current as a consequence of a conductive polymeric film formation. The film thickness was controlled by the number of scans in the electropolymerization process. 75 cycles was considered to be an optimum value, which was obtained after testing the sensor response to solutions of fenitrothion and checking the effectiveness of the template extraction process. | ||
| Fig. 2 Cyclic voltammograms related to the electrochemical synthesis of the polymer in a) absence and b) presence of the template molecule. Measurements recorded with a 5.10−5 M monomer solution in a 0.05 M NaOH medium. | ||
On the other hand, it can be deduced from Fig. 2b, that when the template is added to the polymerization mixture, few changes occur. The anodic and cathodic currents corresponding to the Ni(II)/Ni(III) redox couple continue increasing as the electrosynthesis is taking place but their current intensity values are lower. This could be attributed to the fact that the fenitrothion molecule, once incorporated to the polymeric matrix during the synthesis step, hinders the incorporation of further monomers, resulting in a decrease of the current intensity values of the Ni(II)/Ni(III) redox couple. Furthermore, as has been it was previously suggested by other authors,31,32 the anodic peak at 0.21 V, which disappears in successive scans, may be caused by the oxidation of Ni(II) in Ni(II)-phtalocyanine monomer.
Template extraction
After the electrogeneration of the imprinted polymer, the release of the entrapped template molecule is required in order to leave free the created molecular prints.First of all, it is necessary to corroborate that the fenitrothion has been incorporated to the polymer matrix. This fact, could be deduced by simply measuring the analytical signal provided by the SPCE-MIP through SWV in a Britton-Robinson buffer solution. As can be seen in Fig. 3, a cathodic wave at −0.8 V can be observed in the voltammogram. This signal was attributed to the voltammetric reduction of the template trapped in the polymeric mesh. It may also be noted that, once the template extraction procedure was applied, the reduction wave completely disappeared.
 | ||
| Fig. 3 Square wave voltammograms corresponding to the reduction of the entrapped fenitrothion a) before and b) after the extraction step. Measurements made in a Britton-Robinson 0.04 M buffer solution at pH 1.8. | ||
In order to obtain an efficient method to carry out the template extraction, various strategies are possible: microwave assisted extraction, supercritical fluid desorption, oxidation/reduction of the template in the polymer or, most commonly, the use of a solvent that strongly interacts with the polymer causing the swelling of the coating necessary for template release. In this case, taking into account that SPCE substrate is not compatible with organic solvents, different aqueous buffer solutions were tested at room temperature as well as at 50 °C in order to select the one that best extracted the pesticide. With this objective a series of SPCE-MIPs were synthesized and to each one of them a different extraction procedure was applied. Briefly, the SPCE-MIP surface electrode was submerged in a stirred solution of NaOH (pH = 12.7), a NH4Cl/NH4OH buffer (pH = 9.5) or a Britton Robinson buffer (pH = 3.0), for different periods of time. The obtained results are shown in Fig. 4. As can be observed, as longer extractions were applied, the amount of pesticide entrapped tended to decrease. This effect was more evident when working at 50 °C. The best results were observed when NH4Cl/NH4OH was employed at 50 °C for 15 min.
 | ||
| Fig. 4 Peak intensity values corresponding to the reduction wave of the template entrapped in the polymer after the extraction in a) NaOH 0.05 M, b) Britton Robinson buffer pH 3.0 and c) NH4Cl/NH4OH pH 9.5 buffer, at room temperature or 50 °C. | ||
Voltammetric measurement of fenitrothion with SPCE-MIPs
As mentioned above, the polymeric film of Ni(II)-Phtalocyanine is conductive. This makes it necessary to avoid amperometric signals arising from phenomena of adsorption/diffusion of fenitrothion molecules to the electrode surface. The measurement signal should only come from the fenitrothion attached to the binding sites generated on the polymeric mesh. For this reason it is necessary to carry out an incubation process on the SPCE-MIP device. In order to find out the optimal value of the incubation time, several SPCE-MIP were tested, each one being in contact with a solution containing the target analyte for a fixed period of time. The studied times ranged from 5 to 20 min. No higher analytical signal was obtained even though the sensor was kept for over 10 min in contact with the incubation solution (Fig. 5b). Besides, the effect of the ionic strength during the incubation was studied by increasing the concentration NaCl during the incubation process. The results showed that the peak current of fenitrothion was higher when the concentration of NaCl during the incubation was 1M (Fig. 5a). | ||
| Fig. 5 The influence of a) ionic strength; b) incubation time on the modified SPCE response. Concentration of fenitrothion 5 × 10−5 M. | ||
Once the incubation process was finished, the weakly adsorbed compounds on the modified electrode surface were removed with water and the voltammetric measurement under the above mentioned conditions was performed. Between measurements electrochemical cleaning of the sensor was carried out, through reductive scans between 0 and −1 V on a BR buffer solution at pH 1.8. This brought about a change in the state of oxidation of the template and therefore the interactions between it and the polymeric mesh were weakened. After ten scans the fenitrothion peak had disappeared. Actually, the disappearance of the reductive peak could indicate that the nitro group has been irreversibly reduced to an hydroxylamine group, although it can remain in the polymer mesh. However, there are two experimental facts that corroborate that fenitrothion or its reduced molecule are no longer entrapped in the polymer matrix: first, after the cleaning process the cyclic scans (CV) obtained in a B-R revealed no cathodic or anodic peak and second, the obtained signal after a new incubation with fenitrothion is identical to the first one. Therefore, it could be concluded that after the cleaning step, the fenitrothion and the possible reduced hydroxylamine molecules were removed from their sites.
Periodically, after every 10 determinations, a sweeping of between −1 and 1 V with a scan rate of 200 mV s−1 was applied, in order to clean the electrode surface of possible adsorbed species. This has enabled the electrodic response to be maintained at high levels of reproducibility, without the loss of sensitivity.
Evaluation of the method
The voltammetric determination of fenitrothion using SPCE-MIPs results in well-defined concentration dependences. The linear response found the concentration of fenitrothion to be increasing from 3 × 10−6 to 10−4 M, which follows the equation:| Ip (μA) = (3.1 × 105 ± 0.016 × 105) C (mol L−1) + 0.333 ± 0.078, R2 = 0.9948 |
The LOD (8 × 10−7 M) and the LOQ (3 × 10−6 M) were calculated according to the 3σ and 10σ criteria respectively.
The precision of the method was evaluated studying the repeatability. Ten successive measurements at two concentration levels were used. The RSD obtained were 13.2 and 5.7% for fenitrothion concentrations of 5 × 10−6 M and 10−4 M respectively.
To evaluate the stability of the SPCE-MIPs, ten devices were assayed. Between measurements, SPCE-MIPs were kept in a dried state. The response of devices to 5 × 10−5 M fenitrothion was periodically registered. The current was retained 94% of its initial level after 20 days. Between 20–35 days the devices exhibited 85% of the initial signal and then a fast fall in the response was noted.
A series of different compounds were used to examine the selectivity of the designed MIP sensor. The response of SPCE-MIP was studied using a solution containing the analyte and different potential interfering substances. Experiments were performed by using some structurally related compounds such as the methyl-parathion, fenitrothion-oxon and metamitron. These substances might be present in the same matrix as the fenitrothion and could interfere with its determination.
Fig. 6 shows the molecules studied as possible interferents in the determination of fenitrothion which have reduction peaks on the SPCE close to the reduction peak of fenitrothion (Fig. 6a–6d). When the MIP based screen-printed sensor is incubated with a drop of 40 μL of solutions of each structurally-related pesticide alone, practically no SWV current response can be observed (Fig. 6f–6h). On the other hand, there is no electrochemical response after incubation and cleaning process of the nonimprinted-modified electrode (NIP) in a solution containing fenitrothion and all the studied interferents (Fig. 6e). This fact demonstrates that the resulting imprinted film is highly selective towards the target pesticide and that the observed SWV current responses are not simply due to non-specific adsorption. Finally, when MIP-SPCE is incubated with a mixture of all the interferents including fenitrothion only the peak of the target pesticide can be observed (Fig. 6i–6j). This fact can be attributed to a satisfactory performance with regard to the selectivity of the SPCE-MIP, which is capable of discriminating between molecules with a very similar chemical structure as is the case here with the different pesticides and metabolites.
 | ||
| Fig. 6 Square wave voltammograms showing the reduction peaks of 2 × 10−5 M of a) fenitrothion-oxon, b) metamitron, c) methyl-parathion and d) fenitrothion studied as interferents measured with an unmodified SPCE; e) response after incubation and cleaning process of the nonimprinted-modified electrode (NIP) in a solution containing 2 × 10−5 M of fenitrothion and all the studied interferents. Comparison of the reduction peak obtained with a SPCE-MIP incubated in 2 × 10−5 M solutions of f) fenitrothion-oxon g) metamitron, h) methyl-parathion, i) fenitrothion and j) a solution containing 2 × 10−5 M of fenitrothion and all the interfering compounds. | ||
Analysis of real samples
For the application of the analytical method to real samples, existing data regarding applications of fenitrothion in forest areas was taken into consideration.33 Taking into account the dose of the insecticide applied (0.086 mL m−2), the foliar density (200–300 trees/ha), the percentage of forest area occupied (90%) and the average foliar density of the poplar trees (40 kg leaves per tree), the quantity of pesticide deposited on the forest mass was estimated at 19.35 μg g−1.To simulate the application of the pesticide at two levels of dosage, black poplar leaves were sprayed in the laboratory with Folthion 50 EC obtaining dosages of 20 μg g−1 or 40 μg g−1 in active ingredient. For the fenitrothion extraction 0.5 mL, 1 M NaCl solution was deposed during 10 min over the leave. Longer times did not make any notable improvement to the effectiveness of the in situ extraction and overly increased the analysis times. Following this, 40 μl of the solution was placed on the surface of the SPCE-MIP in order to carry out the incubation process (10 min). After cleaning the electrode with water, the voltammetric measurement was carried out. Ten samples were chosen at random from the treated surface using a previously prepared SPCE-MIP and following the previously described procedure. The obtained recoveries were 24.7 ± 3.6% and 27.5 ± 2.4 for each one of the dosages (Table 1), which may be considered acceptable for a method of these characteristics, of which the main advantage is the portability of the measuring system. The method has been compared with that used by the authors in a previous work,33 based on isotope dilution gas chromatography–mass spectrometry coupled with solid-phase microextraction (SPME/GC-IDMS). The results for the concentration of fenitrothion in leaves are entirely similar, although the SPME/GC-IDMS method shows better RSDs at these concentration levels.
The SPCE-MIP developed method has the advantage of sampling and measuring the concentration of fenitrothion in the studied forest, obtaining in situ results.
Conclusions
A portable and selective SPCE-MIP sensor for the determination of fenitrothion was designed. This aim was achieved by the electropolymerization of Ni(II)-phtalocyanine in the presence of the template molecule on a screen-printed carbon electrode. When the condition for electropolymerization and the step of template extraction were optimized, the sensor exhibited a selective response for fenitrothion in the presence of other possible interferent molecules. When the reduction peak of fenitrothion was studied for solutions at different concentrations, a linear response was found in a range from 3 × 10−6 to 10−4 M, while only a drop of the sample (40 μL) was required. The developed voltammetric sensor with the (hand-held) potentiostat can be used at the sample collection site thereby enhancing rapid detection.Acknowledgements
This work was supported by the Spanish Minister of Science and Innovation (CTQ2008-00651/BQU) and by Commerce and Tourism Department of the Basque Government (Etortek, Project Berrilur IE09-242). The authors would like to thank Javier Alonso for his technical assistance.References
- O. D. Renedo, Talanta, 2007, 73, 202–219 CrossRef.
- J. Wang, TrAC, Trends Anal. Chem., 2002, 21, 226–232 CrossRef CAS.
- C. A. Galan-Vidal and M. E. Paez-Hernandez, in Applications of Analytical Chemistry in Environmental Research, ed. M. Palomar, S. G. Pandalai and A. Gayathri, Research Signpost Trivandrum, Inde, Editon edn, 2005, vol. 2, pp. 23–36 Search PubMed.
- T. Montesinos, S. Perez-Munguia, J.-L. Marty and K. Mitsubayashi, J. Adv. Sci., 2000, 12, 217–222 Search PubMed.
- J. P. Hart, A. Crew, E. Crouch, K. C. Honeychurch and R. M. Pemberton, Anal. Lett., 2005, 37, 789–830 CrossRef.
- A. Mandil and A. Amine, Anal. Lett., 2009, 42, 1245–1257 CrossRef CAS.
- P. Fanjul-Bolado, P. Queipo, P. J. Lamas-Ardisana and A. Costa-Garcia, Talanta, 2007, 74, 427–433 CrossRef CAS.
- M. Angeles Granado Rico, M. Olivares-Marin and E. Pinilla Gil, Talanta, 2009, 80, 631–635 CrossRef CAS.
- P. Fanjul-Bolado, P. J. Lamas-Ardisana, D. Hernandez-Santos and A. Costa-Garcia, Anal. Chim. Acta, 2009, 638, 133–138 CrossRef CAS.
- M. K. Sharma, G. S. Agarwal, V. K. Rao, S. Upadhyay, S. Merwyn, N. Gopalan, G. P. Rai, R. Vijayaraghavan and S. Prakash, Analyst, 2010, 135, 608–614 RSC.
- P. Fanjul-Bolado, P. Queipo, J. Lamas-Ardisana Pedro and A. Costa-Garcia, Talanta, 2007, 74, 427–433 CrossRef CAS.
- M. A. T. Gilmartin, J. P. Hart and B. Birch, Analyst, 1992, 117, 1299–1303 RSC.
- G. Silva Nunes, G. Jeanty and J.-L. Marty, Anal. Chim. Acta, 2004, 523, 107–115 CrossRef CAS.
- M. A. Alonso-Lomillo, O. Dominguez-Renedo, L. Ferreira-Goncalves and M. J. Arcos-Martinez, Biosens. Bioelectron., 2010, 25, 1333–1337 CrossRef CAS.
- M. Yan and O. Ramström, Molecularly Imprinted Materials Science and TechnologyMarcel Decker, New York, 2005 Search PubMed.
- M. C. Blanco-López, S. Gutierrez-Fernandez, M. J. Lobo-Castanon, A. J. Miranda-Ordieres and P. Tunon-Blanco, Anal. Bioanal. Chem., 2004, 378, 1922–1928 CrossRef CAS.
- A. Gómez-Caballero, N. Unceta, M. A. Goicolea and R. J. Barrio, Electroanalysis, 2007, 19, 356–363 CrossRef CAS.
- D. Lakshmi, A. Bossi, J. Whitcombe Michael, I. Chianella, A. Fowler Steven, S. Subrahmanyam, V. Piletska Elena and A. Piletsky Sergey, Anal. Chem., 2009, 81, 3576–3584 CrossRef CAS.
- V. Pichon and F. Chapuis-Hugon, Anal. Chim. Acta, 2008, 622, 48–61 CrossRef CAS.
- K. Mosbach, Trends Biochem. Sci., 1994, 19, 9 CrossRef CAS.
- S. Kröger, A. P. F. Turner, K. Mosbach and K. Haupt, Anal. Chem., 1999, 71, 3698–3702 CrossRef CAS.
- N. Kirsch, J. P. Hart, D. J. Bird, R. W. Luxton and D. V. McCalley, Analyst, 2001, 126, 1936–1941 RSC.
- B. Schöllhorn, C. Maurice, G. Flohic and B. Limogesb, Analyst, 2000, 125, 665–667 RSC.
- T. Panasyuk, V. C. Dall'Orto, G. Marrazza, A. El'skaya, S. Piletsky, I. Rezzano and M. Mascini, Anal. Lett., 1998, 31, 1809–1824 CAS.
- A. Gómez-Caballero, N. Unceta, M. A. Goicolea and R. J. Barrio, Sens. Actuators, B, 2008, 130, 713–722 CrossRef.
- M. C. Blanco-Lopez, M. J. Lobo-Castanon, A. J. Miranda-Ordieres and P. Tunon-Blanco, TrAC, Trends Anal. Chem., 2004, 23, 36–48 CrossRef CAS.
- A. Vasilescu, S. Andreescu, C. Bala, S. C. Litescu, T. Noguer and J.-L. Marty, Biosens. Bioelectron., 2003, 18, 781–790 CrossRef CAS.
- S. de Irazu, N. Unceta, M. C. Sampedro, M. A. Goicolea and R. J. Barrio, Analyst, 2001, 126, 495–500 RSC.
- W. F. Smyth, Polarography of molecules of biological significance, Academic Press, London, 1979 Search PubMed.
- T. Galeano-Díaz, A. Guiberteau-Cabanillas, N. Mora-Díez, P. Parrilla-Vázquez and F. Salinas-López, J. Agric. Food Chem., 2000, 48, 4508–4513 CrossRef CAS.
- M. A. Ruiz, M. G. Blazquez and J. M. Pingarron, Anal. Chim. Acta, 1995, 305, 49–56 CrossRef CAS.
- T.-F. Kang, G.-L. Shen and R.-Q. Yu, Anal. Chim. Acta, 1997, 354, 343–349 CrossRef CAS.
- A. Sánchez, S. Millán, M. C. Sampedro, N. Unceta, E. Rodríguez, M. A. Goicolea and R. J. Barrio, J. Chromatogr., A, 2008, 1177, 170–174 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |