The application of ultrasound for the improvement of analytical procedures: Determination of boron
Jana
Škrlíková
*a,
Vasil
Andruch
*a,
Ioseph S.
Balogh
b,
Hana
Sklenářová
c and
Petr
Solich
c
aDepartment of Analytical Chemistry, University of P.J. Šafárik, SK-04154, Košice, Slovak Republic. E-mail: jana.skrlikova@googlemail.com; vasil.andruch@gmail.com
bDepartment of Chemistry, College of Nyíregyháza, HU-4400, Nyíregyháza, Hungary
cDepartment of Analytical Chemistry, Faculty of Pharmacy, Charles University, CZ-50005, Hradec Králové, Czech Republic
First published on 8th July 2010
Abstract
The aim of the presented study was to investigate the use of ultrasonic irradiation for the improvement of analytical methods. A procedure for boron determining based on the conversion of boric acid to tetrafluoroborate and subsequent spectrophotometric detection, previously reported from our laboratory, was used as a model. The optimum conditions for the conversion of boron to tetrafluoroborate anion (0.9 mol L−1 H2SO4, 0.1 mol L−1 F−, 7 min ultrasonication) as well as for complex formation and extraction of BF4− with DIC (1 × 10−4 mol L−1 DIC) were found. The absorbance of the coloured extracts obeys Beer's law in the range 0.1–1.0 mg L−1 of B. The limit of detection calculated from a blank test (n = 10; P = 0.95) based on 3 s is 0.02 mg L−1 of B. The procedure was applied to the analysis of mineral waters and a pharmaceutical preparative. The main advantage of ultasonification is that the conversion process occurs much faster; reaction time therefore decreases from 30 min to 7 min. Moreover, it does not lead to the heating of the sample, meaning, as a consequence, the value of the blank test decreases, and the reaction does not have to be performed in a closed system.
Introduction
Ultrasound is a well-known and widely used auxiliary energy which is frequently used in a variety of fields as a substitute for heating or microwave irradiation. Analytical chemistry is one field in which ultrasound has not yet been exploited, although this auxiliary technique could be a powerful aid in improving various analytical procedures. Ultrasonic energy has been applied in analytical chemistry mainly for (a) pre-treatment of solid samples (extraction, homogenisation);1 (b) ultrasonic-assisted derivatisation reactions, mainly in gas chromatography;2 (c) various other operations, such as nebulisation in atomic spectrometry, glassware cleaning and the removal of bubbles from solution; as well as (d) detection.3 Here it is necessary to underline the works of Capote and de Castro, in particular, their review of ultrasound use.4 It is also especially important to note the numerous works of Coakley and co-workers devoted primarily to the application of ultrasound in biology and medicine for particle/cell separation5–11 as well as their review, in which applications of ultrasound streaming and radiation force in biosensors were described.6Although many articles have examined the application of ultrasound for the above-mentioned purposes, the number of articles devoted to investigating this energy in order to improve analytical reactions is limited. De Sousa and Korn investigated the influence of ultrasonic waves in the spectrophotometric determination of nickel with dimethylglyoxime12 and showed that the nickel determination could be performed without the use of masking agents and oxidising solutions. They demonstrated that generating free radicals by exposing aqueous solutions to ultrasonic waves is a means of improving both the sensitivity and selectivity of spectrophotometric methods of analysis. Implementation of ultrasonic irradiation into analytical procedures in general and replacing heating with ultrasonication in particular can offer a series of advantages, some of which have already been indicated in the literature,12 such as improving the analytical characteristics of determinations, the possibility of automation and thus a decrease in operator handling, and reducing the possibility of sample and environmental contamination as well. Ultrasonication can lead to the acceleration of chemical reactions and the subsequent shortening of analysis time, which could possibly be explained by the different functional mechanism of ultrasonic irradiation on the sample. The main advantage of using ultrasonic energy instead of heat lies in the different effect these two forms of auxiliary energy have on an aqueous sample. The chemical effect of ultrasonic waves probably originates from cavitations, which favour the diffusivity of the dissolved particles in the sample, as well as from the oxidative properties of the radicals formed during ultrasonification. It is important to emphasize that the formation of bubbles subjected to fast adiabatic compressions and expansions gives rise to high temperatures and pressures inside them, though these fluctuations have minimal effect on the temperature of the system as a whole.13
We previously reported a method for boron determination based on the reaction of boric acid with fluoride upon heating and the formation of tetrafluoroborate anion, followed by the extraction of BF4− with Victoria blue 4R reagent into benzene and subsequent spectrophotometric detection.14 The conversion of boric acid to HBF4 was relatively slow, however. Heating significantly speeds up HBF4 formation. The primary difficulty in this reaction is the necessity of heating at 70 °C for a relatively long period (30 min) in a closed system in order to prevent the release of the volatile products formed. This step is performed in closed polyethylene test tubes. The heating must be continuous to ensure quantitative conversion, but at the same time very gentle in order to prevent deformation of the polyethylene test tubes as a result of increasing pressure in the closed system.
The aim of the presented study was to investigate the feasibility of using ultrasound instead of heating during the conversion of boric acid to tetrafluoroborate in order to accelerate and improve the analytical procedure, and thus the development of a novel spectrophotometric method for boron determination. The main advantage of ultasonification is that it does not lead to the heating of the sample, meaning, as a consequence, that the value of the blank test decreases and the reaction does not have to be performed in a closed system. Moreover, the conversion process is much faster; therefore the reaction time decreases from 30 to 7 min.
Experimental
Reagents
All chemicals and solvents used were of analytical grade purity (unless stated otherwise). Ultra pure water produced by a Millipore Milli-Q RG system (Millipore, USA) was used throughout the work. All solutions were stored in polyethylene bottles. A stock solution containing 1 × 10−2 mol L−1 of boron was prepared by dissolution of 0.0618 g H3BO3 in water and dilution with water up to a volume of 100 mL. A 1 × 10−3 mol L−1 boron working solution was prepared by dilution of the stock solution. A 1 × 10−3 mol L−1 aqueous solution of dimethylindocarbocyanine dye (DIC) was prepared by dissolving 0.0195 g of its chloride salt in 1 mL of methanol (p.a.) and subsequent dilution with water up to a volume of 50 mL. A 0.5 mol L−1 aqueous solution of NaF was used to convert H3BO3 to BF4−. The pH was set by the addition of 3 mol L−1 H2SO4.Apparatus
A diode array UV-Visible Spectrophotometer 8453 (Agilent, USA) and a Spekol-11 spectrophotometer (Carl Zeiss, Germany) equipped with a matched cell of 10 mm path length were used for spectrophotometric measurements. A Sonorex Digitec ultrasonic compact bath DT 31 equipped with a HF generator with constant power 30 Weff. (Bandelin Electronic, Germany) and a UZD 500 Ultrasound disintegrator (Person-Ultragen, Slovakia) equipped with a cylindrical titanium alloy probe and having an adjustable ultrasonic generator with a maximal power of 500 W were used as a sources of ultrasonic energy for the conversion of boric acid to tetrafluoroborate anion.Calibration of the method
Aliquots (up to 0.5 mL) of the H3BO3 working solution (1 × 10−3 mol L−1) were introduced into 10 mL polyethylene test tubes, then 0.2 mL of 0.5 mol L−1 NaF and 0.3 mL of 3 mol L−1 H2SO4 were added and the volume was filled up to the 1 mL with water. After the addition of each reagent, the solution was mixed thoroughly. The solution was then sonicated in an ultrasonic bath for 7 min. Approx. 10–15 test tubes can be sonicated simultaneously. Next, 3.5 mL water and 0.5 mL of 1 × 10−3 mol L−1 DIC were added, and the solution was mixed well. The complex was then extracted using 5 mL of amyl acetate for 60 s. The absorbance was measured at a wavelength of 551 nm in a matched cell of 10 mm path length against a blank test.Procedure for real samples
An aliquot of sample of up to 0.5 mL, depending on the expected boron content, was introduced into a polyethylene test tube. All required reagents were then added and the determination performed as described in the Calibration of the method section.Results and discussion
Since the conversion of boric acid to HBF4 is relatively slow, in previous work14 we used heating to accelerate the reaction. Although the reaction of boric acid with fluoride is well-known,15,16 we found no information in the literature about the effect of ultrasonic energy on this reaction. Therefore, we studied various physical factors, as well as the chemical conditions, which can influence the conversion process.Investigation of ultrasonic energy on the conversion of H3BO3 to BF4−
Two different devices were used as sources of ultrasonic energy: an ultrasonic bath with constant power and an ultrasonic disintegrator with energy adjustable in the range of 200–500 W. Effects of energy, ultrasonication time and experimental conditions (acidity of the medium and fluoride concentration) were investigated.Effect of energy
The effect of ultrasonic energy was investigated using an ultrasonic probe (Fig. 1). For this purpose, a series of experiments were carried out with a constant concentration of boric acid (6 × 10−5 mol L−1) and in the presence of all reagents necessary for the conversion process (0.9 mol L−1 H2SO4 and 0.1 mol L−1 F−) at the selected sonication time of 1 min. The ultrasonic probe was immersed in a water bath in which the extraction test tubes were placed. After sonication, the appropriate amounts of water and dye reagent were added to the test tubes, and the determination followed by extraction using 5 mL of amyl acetate and spectrophotometric detection, as described in the Calibration procedure section. Increasing the energy did not significantly influence the value of the analytical signal, but it did cause an undesirable increase in the blank tests. Therefore 250 W can be recommended for the conversion process.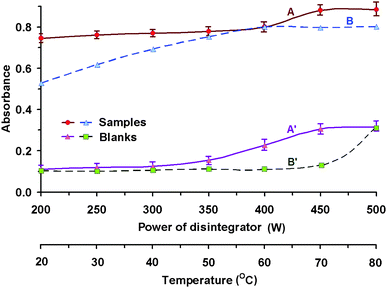 | ||
| Fig. 1 Effect of the energy of the ultrasonic disintegrator conditions for conversion: 6 × 10−5 mol L−1 H3BO3; 0.9 mol L−1 H2SO4; 0.1 mol L−1 F−; Conditions for extraction: 1 × 10−4 mol L−1 DIC; 5 mL amyl acetate; l = 10 mm; λ = 551 nm; A, A′ – ultrasonic disintegrator (1 min); B, B′ – conventional water bath heating (30 min); A, B – complexes; A′, B′ – blank tests. | ||
Besides the effect of ultrasonic energy, Fig. 1 also shows the influence of temperature on the conversion process (the broken lines). As we can see, no significant differences in absorbance were found when using two different types of auxiliary energy. The advantage of using ultrasonic energy in comparison with heating is the significant decrease in analysis time.
Effect of ultrasonication time
The time of ultrasonication was investigated within a range from 1 to 40 min (Fig. 2). Comparing results from the two different devices, we found that the maximum analytical signal was consistently achieved in a shorter time of just over 1 min with the use of the ultrasonic probe. To achieve a comparable signal, approx. 5–8 min and 30 min were needed in the case of the ultrasonic bath and conventional heating, respectively. But no significant difference was observed in the value of analytical signal obtained. It is true that the ultrasonic probe seems to be more advantageous due to the shorter analysis time, but despite this fact, we recommend using an ultrasonic bath because of the lower cost of the equipment and higher accessibility in common analytical laboratories. The time selected for further study was 7 min (ultrasonic bath). With a shorter time, the conversion was insufficient, while with an increase in time, the efficiency of the ultrasonic energy for conversion remained constant. With longer times, however, increasing the temperature of the water in the ultrasonic bath leads to an increase in the absorbance of the blank test. Therefore, the water in the ultrasonic bath needs to be changed after every third sonication. It is also possible to connect the ultrasonic bath to a thermostat in order to maintain a constant temperature of 20 °C.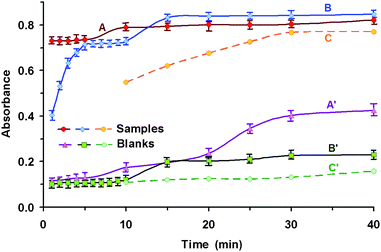 | ||
| Fig. 2 Effect of the time of ultrasonication on the conversion of H3BO3 to tetraflouroborate conditions for conversion: 6 × 10−5 mol L−1 H3BO3; 0.9 mol L−1 H2SO4; 0.1 mol L−1 F−; Conditions for extraction: 1 × 10−4 mol L−1 DIC; 5 mL amyl acetate; l = 10 mm; λ = 551 nm; A, A′ – ultrasonic disintegrator (250 W); B, B′ – ultrasonic bath; C, C′ – conventional water bath heating (70 °C); A, B, C – complexes; A′, B′, C′ – blank tests. | ||
Effect of experimental conditions
We studied the experimental conditions for the conversion of H3BO3 to tetrafluoroborate in a previous work14 and determined that for complete conversion, the medium must be sufficiently acidic to suppress hydrolysis and that there must be an adequate surplus of fluoride anion present. However, since heating is now being replaced by ultrasonic energy, the appropriate experimental conditions must again be specified. Therefore, a series of experiments were carried out (at the selected sonication time of 7 min) with a constant concentration of boric acid (6 × 10−5 mol L−1) and varying concentrations of sulfuric acid and fluoride ions (Fig. 3). The appropriate conditions were found to be: 0.9 mol L−1 H2SO4 and 0.1 mol L−1 NaF. These conditions differ significantly from than those obtained in7 (0.1 mol L−1 H2SO4, 0.02 mol L−1 NaF; heating time 30 min; temperature 70 °C).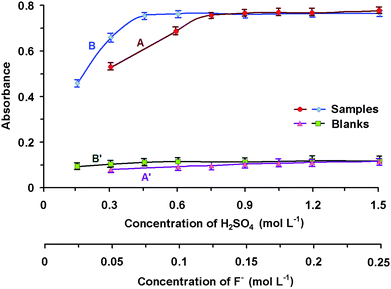 | ||
| Fig. 3 Effect of concentration of the of sulfuric acid (A, A′) and fluoride ions (B, B′) on the conversion of H3BO3 to tetraflouroborate Conditions for conversion: 6 × 10−5 mol L−1 H3BO3; 7 min sonication in an ultrasonic bath; 1,1′: 0.1 mol L−1 F−; 2,2′: 0.9 mol L−1 H2SO4; Conditions for extraction: 1 × 10−4 mol L−1 DIC; 5 mL amyl acetate; l = 10 mm; λ = 551 nm; A, B – complexes, A′, B′ – blank tests. | ||
Investigation of the extraction process
The following factors can affect the formation and extraction of the boron complex: the pH of the aqueous phase, the concentration of DIC and the nature of the organic solvent. The main problem in the procedure for determining boron based on the conversion of H3BO3 to tetrafluoroborate anion and subsequent complex formation with dye reagents arises from the high acidity of the medium (0.9 mol L−1 H2SO4) required for complete conversion. On the other hand, according to the reaction mechanism (see below), the dye reagent has to be in a single charged form. Most dye reagents, however, are more or less protonated at such an acidity and are not able to form complexes with BF4− that are extractable by organic solvents. Therefore, an additional neutralisation step is needed between conversion and complex formation.14 In contrast to Ref. 14, in this work dimethylindocarbocyanine dye (Fig. 4) was used as the complexing reagent. Based on the investigation of the acid–base properties as well as spectrophotometric characteristics of DIC17 we can say that DIC exists in single-charged form over a wide range of acidity. This allows for the use of DIC for boron complex formation without neutralization of the solution after conversion, which is an advantage of the suggested procedure. The appropriate concentration of dye reagent was found to be: 0.8–1.4 × 10−4 mol L−1.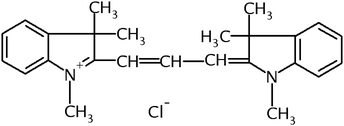 | ||
| Fig. 4 Structural formula of DIC. | ||
Various groups of organic solvents, such as aliphatic and aromatic hydrocarbons, higher alcohols, ketones, acetic esters and the halogen and nitrogen derivatives of aliphatic and aromatic hydrocarbons were tested as extractants. The best extractants appeared to be benzene and amyl acetate, as both of them showed very good extractability of the complex and minimum extractability of the dye reagent. Amyl acetate, which is less toxic than benzene, was chosen as the organic solvent for the extraction of the boron complex. The extraction equilibrium is reached within 40–60 s, and the absorbance of the extracts remains stable for several hours.
Reaction mechanism
The determination procedure involves two independent steps: (1) conversion of boric acid to tetrafluoroborate anion, and (2) complex formation and extraction of BF4− formed with a cation of DIC. The molar ratio of BF4− and DIC in the extracted complex determined under the appropriate experimental conditions using various spectrophotometric methods (Job's method, Asmus' method, and the relative yield method) was found to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Based on these results, it can be assumed that the reaction mechanism may be expressed by the following chemical equations:
:1. Based on these results, it can be assumed that the reaction mechanism may be expressed by the following chemical equations:| H3BO3 + 4F− + 3H+ = BF4− + 3 H2O |
| BF4−(aq) + (DIC)+(aq) + nS(org) = [BF4]−(DIC)+ × nS(org) |
Method validation
| a A = a + bC, where A means the absorbance, a the intercept, b the slope and C the concentration of B in mg L−1; n = 10, P = 0.95. | |
|---|---|
| Wavelength/nm | 551 |
| Extractant | Amyl acetate |
| Regression equationa | |
| Intercept, a | 0.0027 ± 0.0011 |
| Slope, b | 0.9497 ± 0.0189 |
| Correlation coefficient, r | 0.9989 |
| Concentration range/mg L−1 | 0.1–1.0 |
| LOD/mg L−1 | 0.02 |
| Taken/mg L−1 | Intra-day | Inter-day | ||||
|---|---|---|---|---|---|---|
| Determineda/mg L−1 | RSD (%) | R (%) | Determineda/mg L−1 | RSD (%) | R (%) | |
a
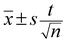 t – Student coefficient for n − 1 degrees of freedom.
t – Student coefficient for n − 1 degrees of freedom.
|
||||||
| 0.20 | 0.21 ± 0.01 | 4.5 | 105.0 | 0.19 ± 0.01 | 5.0 | 95.0 |
| 0.35 | 0.34 ± 0.01 | 2.8 | 97.1 | 0.36 ± 0.01 | 2.6 | 102.9 |
| 0.65 | 0.66 ± 0.01 | 1.4 | 101.5 | 0.66 ± 0.02 | 2.9 | 101.5 |
| 0.80 | 0.78 ± 0.02 | 2.4 | 97.5 | 0.83 ± 0.03 | 3.4 | 103.8 |
The obtained results given in Table 3 give evidence of the good reproducibility of the developed procedure, since RSD did not exceed 3.5%. The robustness of the procedure was assessed by evaluating the influence of small variations in the experimental variables: the concentration of fluoride ions, sulfuric acid, and dye as well as the extraction time. In this study, one parameter was changed while the other parameters were kept constant. The percentage of relative standard deviation and recovery percentage were then calculated for each variation. No small variations in any of the variables significantly affected the results. The RSD was in all cases less than 2.1%, and the recovery was in the range of 99.5–103.3%.
| Sample | Taken/added/mg L−1 | Instrument I. | Instrument II. | ||||
|---|---|---|---|---|---|---|---|
| Determined/mg L−1 | RSD (%) | R (%) | Determined/mg L−1 | RSD (%) | R (%) | ||
| Internal | 0.28 | 0.28 ± 0.01 | 3.4 | 100.0 | 0.27 ± 0.01 | 3.5 | 96.4 |
| standard | 0.48 | 0.48 ± 0.01 | 2.0 | 100.0 | 0.48 ± 0.01 | 2.0 | 100.0 |
| solution of H3BO3 | 0.76 | 0.76 ± 0.01 | 1.3 | 100.0 | 0.77 ± 0.01 | 1.2 | 101.3 |
| 0.92 | 0.91 ± 0.01 | 1.0 | 98.9 | 0.92 ± 0.01 | 1.0 | 100.0 | |
| NW I. | 0.36 | 0.35 ± 0.01 | 2.7 | 97.2 | 0.37 ± 0.01 | 2.6 | 102.8 |
| 0.56 | 0.56 ± 0.02 | 3.4 | 100.0 | 0.57 ± 0.01 | 1.7 | 101.8 | |
| NW II. | 0.26 | 0.27 ± 0.01 | 3.5 | 103.8 | 0.27 ± 0.01 | 3.5 | 103.8 |
| 0.70 | 0.69 ± 0.01 | 1.4 | 98.6 | 0.72 ± 0.01 | 1.3 | 102.9 | |
:interferent ratio that caused an error not higher or lower than ± 5%. About a 2000-fold excess of CH3COO−, Cl−, SO42−, Ca2+, K+, Na+, a 1000-fold excess of Mg2+, a 500-fold excess of Cu2+, a 300-fold excess of Fe2+ and a 250-fold excess of Ni2+ did not interfere with the determination of boron. Some ions, such as NO3−, Br− and I−, easily formed a complex with DIC and therefore strongly interfered with boron determination. These ions must therefore be separated before conversion of boron to BF4− using the multiple extraction procedure previously described in our work.14 Boron can then be determined according to the procedure suggested in this work. In this case, about a 100-fold excess of NO3− and I− and a 150-fold excess of Br− did not interfere the determination of boron.
The suggested method was also applied to the analysis of seawater samples spiked with two different amounts of boron (0.48 and 0.92 mg L−1). The determined amounts of boron found were 0.47 ± 0.01 mg L−1 and 0.91 ± 0.01 mg L−1 with a precision of 0.25–0.50% RSD.
Acknowledgements
H.S. and P.S. would like to thank to the Ministry of Education of the Czech Republic for its financial support (MSM 0021620822). J.S. would like to say a special thanks to the International Visegrad Fund for providing a 10-month Scholarship and internal grant system of Faculty of Science of Pavol Jozef Šafárik University in Košice (VVGS Grant No. PF 4/2010/Ch).References
- A. Caballo-López and M. D. Luque de Castro, J. Chromatogr. A, 2003, 998, 51 CrossRef CAS.
- Y. C. Fiamegos, C. G. Nanos and C. D. Stalikas, J. Chromatogr. B, 2004, 813, 89 CrossRef CAS.
- W. Lok (Jacky) Chou and P. C. H. Li, Anal. Chim. Acta, 2006, 572, 39 CrossRef.
- F. Priego Capote and M. D. Luque de Castro, Anal. Bioanal. Chem., 2007, 387, 249.
- W. T. Coakley, J. J. Hawkes, M. A. Sobanski, C. M. Cousins and J. Spengler, Ultrasonics, 2000, 38, 638 CrossRef CAS.
- L. A. Kuznetsova and W. T. Coakley, Biosens. Bioelectron., 2007, 22, 1567 CrossRef CAS.
- M. S. Limaye, J. J. Hawkes and W. T. Coakley, J. Microbiol. Methods, 1996, 27, 211 CrossRef.
- W. T. Coakley, D. Bazou, J. Morgan, G. A. Foster, C. W. Archer, K. Powell, K. A. J. Borthwick, C. Twomey and J. Bishop, Colloids Surf. B: Biointerfaces, 2004, 34, 221 CrossRef CAS.
- C. M. Cousins, P. Holownia, J. J. Hawkes, C. P. Price, P. Keay and W. T. Coakley, Ultrasonics, 2000, 38, 654 CrossRef CAS.
- K. A. J. Borthwick, W. T. Coakley, M. B. McDonnell, H. Nowotny, E. Benes and M. Grfschl, J. Microbiol. Methods, 2005, 60, 207 CrossRef CAS.
- N. R. Harris, M. Hill, S. Beeby, Y. Shen, N. M. White, J. J. Hawkes and W. T. Coakley, Sens. Actuators B, 2003, 95, 425 CrossRef.
- C. Santos de Sousa and M. Korn, Anal. Chim. Acta, 2001, 444, 309 CrossRef.
- R. Japón-Luján, J. M. Luque-Rodríguez and M. D. Luque de Castro, J. Chromatogr. A, 2006, 1108, 76 CrossRef CAS.
- I. S. Balogh, V. Andruch, M. Kádár, F. Billes, J. Posta and E. Szabová, Int. J. Environ. Anal. Chem., 2009, 89, 449 CrossRef CAS.
- A. A. Nemodruk and Z. K. Karalova, in Boron (in Russian), ed. Nauka, Moscow, 1964, p. 86 Search PubMed.
- J. Katagiri, T. Yoshioka and T. Mizoguchi, Anal. Chim. Acta, 2006, 570, 65 CrossRef CAS.
- I. S. Balog, P. P. Kish, A. A. Ishchenko, I. L. Mushkalo and V. A. Andrukh, J. Anal. Chem., 1990, 45, 344.
- ČSN ISO 9390, Water Quality – Determination of borate. Spectrophotometric determination with azomethine- H (in Czech), Czech normalization institute, 1996 Search PubMed.
| This journal is © The Royal Society of Chemistry 2010 |