On-line UV photoreduction in a flow-injection/stopped-flow manifold for determination of mercury by cold vapour-atomic absorption spectrometry
Sandra
Gil
,
Marta
Costas
,
Franciso
Pena
,
Inmaculada
De La Calle
,
Noelia
Cabaleiro
,
Isela
Lavilla
and
Carlos
Bendicho
*
Departamento de Química Analítica y Alimentaria, Área de Química Analítica, Facultad de Química, Universidad de Vigo, Campus As Lagoas-Marcosende s/n, 36310, Vigo, Spain. E-mail: bendicho@uvigo.es; Fax: +34-986-812556; Tel: +34-986-812281
First published on 24th September 2010
Abstract
Photo-chemical vapour generation has been applied in a flow-injection system under stopped-flow conditions for determination of Hg by atomic absorption spectrometry. The system allows mercury vapour generation without the need for conventional reduction reactions based on sodium/potassium tetrahydroborate (III) or tin chloride in acid medium. The photo-induced reaction is accomplished by applying ultraviolet irradiation (UV) to the sample solution containing Hg(II) in the presence of an organic acid (i.e., acetic, citric, oxalic, ethylendiaminetetraacetic) as precursor of reducing species. A remarkable improvement in sensitivity is observed with acetic acid when stopped-flow is employed as compared to continuous operation, meaning that kinetics play an important role in the photo-induced reaction. A detection limit of 0.3 μg L−1 can be obtained, which represents a 6-fold improvement in respect to that obtained without stopped-flow. The repeatability expressed as relative standard deviation was about 2.7% (n = 15) for a 50 μg L−1 Hg standard. The effect of potential interferences on the photo-generation of Hg vapour was investigated. In the UV-photo-induced CVG, both inorganic Hg and organomercury species can be reduced to elemental mercury with the same efficiency. The method was applied to determination of Hg in several enriched natural water samples and recoveries of MeHg+, Thiomersal, EtHg+ and PhHg+ were in the range 97% to 102%.
Introduction
Mercury pollution is recognized as a primary environmental issue and public health problem.1 A great variety of industrial uses involve Hg and its compounds, e.g., as catalysts in the chloralkali industry, preservatives and pigments in the paint industry, pesticides, pulp and paper industry, battery production, etc.2 Additionally, toxicity of Hg can be increased upon transformations occurring in the environment.3 Many international guidelines have limited maximum allowable contents of Hg in water and food. Thus, the U.S. environmental protection agency (EPA) has limited Hg concentration in water for human consumption to 2 μg L−1 in accordance with the primary drinking water standard,4 while the European Community has indicated a maximum Hg concentration of 1 μg L−1 in drinking water.5 Owing to the extremely low concentration of Hg in natural water, environmental and biological samples, very sensitive analytical methods are often required for accurate determination.The most used technique worldwide for the determination of mercury is cold vapour generation (CVG) with detection by atomic absorption spectrometry (AAS) or atomic fluorescence spectrometry (AFS),6 because of its simplicity, high sensitivity, and freedom from interferences. Hg(II) can be effectively converted into Hg(0) by chemical reductants such as stannous chloride or sodium tetrahydroborate (III), and subsequently swept by a carrier gas into the detection system for determination. Main disadvantages include chemical interferences from transition metals, unstable reductant solution, vigorous chemical reactions that can result in liquid transport to the atomization cell and significant waste production.
Development of new vapour generation systems that may replace or diminish the use of chemical reagents remains an attractive research topic.7 An alternative to chemical reduction is the use of photoreduction, based on the exposure of a sample to UV radiation.8 Photo-CVG may provide a powerful alternative to conventional CVG owing to its simplicity, versatility, and cost-effectiveness. Photo-induced CVG eliminates the need for freshly prepared chemical reductant, can simplify the flow-injection manifold by removing a reductant line and should lead to a smoother, less violent reduction process.
Photo-induced CVG can be carried out by exposure of Hg(II) (and MeHg+) solutions to UV irradiation in the presence of low molecular weight organic acids with or without a photocatalyst such as TiO2,9–17 although other precursors such as mercaptoethanol,18 ethanol,19 aldehydes,20etc have also been tried. So far, UV photoreduction for total Hg determination has been performed in batch and flow systems coupled to a variety of detection systems such as atomic fluorescence spectrometry,10,16–20 inductively-coupled plasma mass spectrometry,9,15 quartz tube-atomic absorption spectrometry11,13,14 and graphite furnace-atomic absorption spectrometry.12 Advantages of flow systems include automation and increased sample throughput. So far, continuous operation has been mostly employed in flow systems for photo-CVG of mercury vapours but flow injection has been scarcely investigated. Regarding the latter approach, It is clear that the residence time of the sample zone in the flow-through photoreactor should largely influence the sensitivity and detection limit achieved, since the dose of UV radiation received determines the extent of radical formation and, in turn, the efficiency of UV reduction ionic Hg species.
In earlier work, the authors have demonstrated the feasibility of on-line manifolds with flow-injection/stopped-flow (FI/SF) operation for photo-CVG of Se vapours.21
In this work, UV irradiation of Hg(II) solutions containing several organic acids (i.e., acetic, oxalic, citric and ethylendiaminetetraacetic) as precursors of reducing species was investigated for the determination of Hg by FI/SF-photo-CVG-AAS. Stopped-flow conditions were employed with the aim of increasing the time for which the sample zone was subjected to UV irradiation while being transported to the detector.
Experimental
Apparatus
A Thermo Electron Corporation® series M5 atomic absorption spectrometry (Cambridge, UK) equipped with deuterium background corrector and a quartz cell located in the optical path was employed. A wavelength of 253.7 nm and a spectra bandpass of 0.5 nm were employed. Volatile mercury was generated using the system shown in Fig. 1. The photo-induced reduction process was accomplished with a flow-through photoreactor (Model 10.570) from PS Analytical (UK), consisting of a 12 m PTFE tubing of dimensions 0.8 mm i.d. × 1.2 mm o.d. wrapped around a low-pressure Hg lamp (78 W power). The organic acid carrier stream was pumped through the photoreactor by a Gilson Model Minipuls 3M peristaltic pump. Flow injection operation was performed with a Chemiert model C12 eight-port external volume sample injector (Valco instruments Co., Houston, USA) incorporating a 200 μL capacity loop. Separation of volatile compounds was performed with a Perkin-Elmer PTFE membrane gas-liquid separator (Überlingen, Germany). High purity Ar was employed to carry the Hg vapour from the gas-liquid separator to the quartz cell. The Ar flow-rate was controlled by a flow-meter from Fisher and Porter (Warminster, PA). | ||
| Fig. 1 Schematic diagram of the flow-injection/stopped-flow manifold including a flow-through photoreactor for on-line generation of Hg vapour. | ||
Reagents
All chemicals were of analytical-reagent grade. A Hg(II) stock standard solution (1000 mg L−1) was obtained from Hg(NO3)2 (Panreac). Ultrapure water from a PETLAB water purifier system (Peter Taboada) was used. Thiomersal (i.e., ethylmercurithiosalicylate) (Sigma), methyl-, ethyl- and phenyl-mercury chlorides (Riedel de Haën) were used to evaluate the FI/SF-photo-CVG-AAS system for organomercurials. A stock solution of 500 mg L−1 thiomersal was prepared in ultrapure water. Stock solutions of MeHg+, EtHg+ and PhHg+ were prepared in methanol–water mixture at 500 mg L−1 (as Hg) concentration. All stock solutions were protected against light and stored at 4 °C in the dark. The following chemicals were employed for the interference study: NaCl, Na2CO3, CaCl2, MgCl2·6H2O, Mg(NO3)2·6H2O, Fe(NO3)3·9H2O, CuCl2·2H2O, CrCl3·6H2O from Merck; KCl from Prolabo; KNO3 and NiCl2 from Probus; MnCl2·4H2O and CoCl2·6H2O from Scharlau; Mn(NO3)2·4H2O, Ni(NO3)2·6H2O, Cd(NO3)2·4H2O, K2CrO4, and Pb(NO3)2 from Panreac; Cr(NO3)3 and CaCO3 from Sigma-Aldrich; humic acid from Fluka Chemica. Organic acids used like precursors of reducing species in the photogeneration of Hg vapours were the following: acetic, citric and oxalic acids from Merck and ethylenediaminetetraacetic acid from Prolabo. Other chemicals used were K2Cr2O7, NaOH, HCl, and HNO3 (Prolabo). High purity Ar was employed to sweep out the Hg vapour to the quartz cell.Procedure
A schematic diagram of the FI/SF-photo-CVG-AAS system is depicted in Fig. 1. Determination of Hg by FI/SF-photo-CVG-AAS was carried out according to the following procedure: the carrier was constantly pumped through the flow-through photoreactor and a 200 μL volume of sample was loaded in the injection valve to fill the loop. In the ‘inject’ position, the sample was introduced into the carrier stream. The sample zone was arrested inside the flow-through photoreactor after the sample injection for a variable time depending on the organic acid employed. An Ar stream flushed the Hg vapour into the gas-liquid separation unit, which was connected to the quartz cell by PTFE transfer tubing.Wastes were withdrawn from the gas-liquid separator using the same peristaltic pump. Atomic absorption profiles could be integrated within 100 s. The atomic absorption signal was recorded and the peak height was used for quantification. Spiked natural freshwater were made in order to evaluate the proposed method. The final concentration in the samples was 50 μg L−1 Hg.
Water samples (except mineral water and CRM NWTM-27.2) were filtrated through 0.45 μm cellulose nitrate filters (Sartorius) and stored at 4 °C.
Optimal conditions using acetic acid as carrier were the following: a 16.5 mL min−1 acid carrier stream flow-rate, a 100 mL min−1 argon flow-rate, a 150 s stopped-flow time and a 2.5 mol L−1 acetic acid concentration.
Results and discussion
Effect of instrumental parameters and reaction conditions on UV-photo-induced cold vapor generation
When applying on-line treatments in flow-injection manifolds, stopped-flow methods can be implemented so that kinetically slow reactions can be used. Stopped-flow conditions allow the sample zone to be UV-irradiated for the required time with minimum dispersion. All optimization studies were performed with a 50 or 100 μg L−1 Hg(II) aqueous standard.The stopped-flow time was studied between 0 and 300 s. Results showed that improved sensitivity is generally reached using stopped-flow (Fig. 2). Stopped-flow times of 120, 150 and 240 s are needed to obtain maximum Hg absorbance with oxalic, acetic and ethylendiaminetetraacetic acids, respectively. No effect is observed with citric acid when stopped-flow is implemented. The most outstanding effect is observed with acetic acid. Peak absorbance is increased about six times as compared to experiments without stopped-flow. However, slight improvement is observed with the rest of the acids. Photoreduction kinetics have not been taken into account in previous work with the acetic acid system regarding the photogeneration of Hg vapour in on-line systems, but it is clear from these results that a noticeable enhancement in absorbance is achieved when the sample zone is subjected to longer UV irradiation times. If the manifold is employed in the flow-injection mode without stopped-flow and with lower flow-rates for the carrier in an attempt to increase the residence time, wide atomic absorption signals are obtained, which are difficult to integrate within the measurement time. So, sensitivity is drastically decreased. This is due to the increased dispersion occurring in such manifold, which counteracts the positive effect of the increased residence time of the sample zone inside the photoreactor.
 | ||
| Fig. 2 Effect of the stopped-flow time on the Hg absorbance using different organic acids precursors for photogeneration of Hg vapour. (50 μg L−1 Hg for all acids; the concentration of each acid was: acetic, 1.2 mol L−1; EDTA, 0.125 mol L−1; citric, 0.4 mol L−1; oxalic, 0.4 mol L−1). | ||
The influence of the acid concentration is shown in Fig. 3. Differences between optimal acid concentrations required with each acid are evident. While a maximum is reached with ca. 2.5 mol L−1 for acetic acid, much smaller concentrations were required with citric, oxalic and EDTA. It is remarkable to observe that only 0.05 mol L−1 of EDTA, 0.2 mol L−1 of citric and 0.2 mol L−1 of oxalic acid concentration are needed to obtain an optimum photoreduction efficiency, although poor precision is observed for the above acids when low acid concentration is used.
 | ||
| Fig. 3 Effect of organic acid concentration used as carrier on the Hg absorbance following photogeneration of Hg vapour (50 μg L−1 for acetic acid and 100 μg L−1 for the remaining acids. The stopped-flow times employed with each acid were: acetic, 150 s; EDTA, 240 s; citric, no stopped-flow; oxalic, 120 s). | ||
The effect of the quartz cell temperature on the Hg absorbance when the photoreduction is performed in acetic acid medium was studied in the range 25-900 °C (Fig. 4). Best results were obtained without heating the quartz cell, meaning that Hg(0) was the Hg species reaching the measurement cell.

As can be observed in Fig. 5, the effect of Ar flow-rate on peak absorbance was found to be critical. The Ar flow-rate was varied between 40 and 100 mL min−1. The flow-rate chosen for further experiments was 100 mL min−1. Low flow-rates provided smaller and wider peaks that could not be integrated within the measurement time.
 | ||
| Fig. 5 Effect of the Ar flow-rate on Hg absorbance. | ||
Effect of foreign substances
Under stopped-flow conditions, the effects of several coexisting ions and humic acid on the photogeneration of Hg vapour with the acetic acid system was investigated. The effect of potential interferents over the absorbance corresponding to a 50 μg L−1 Hg(II) solution can be observed in Table 1. An interference effect is considered to be significant when it is beyond ±10%.| Interferent | Interferent concentration/mg L−1 | Effect (%) |
|---|---|---|
| Na2CO3 | 100 | −24.89 |
| NaCl | 100 | +12.06 |
| KNO3 | 100 | +6.99 |
| KCl | 100 | +15.13 |
| Mg(NO3)2·6H2O | 100 | +0.90 |
| MgCl2·6H2O | 100 | +20.63 |
| CaCO3 | 100 | +17.52 |
| CaCl2 | 100 | +16.26 |
| CrCl3·6H2O | 10 | +4.72 |
| Cr(NO3)3 | 10 | +0.89 |
| K2CrO4 | 10 | −4.00 |
| MnCl2·4H2O | 10 | +7.16 |
| Mn(NO3)2·4H2O | 10 | +6.20 |
| CoCl2·6H2O | 10 | −22.42 |
| Cd(NO3)2·4H2O | 10 | −0.82 |
| Pb(NO3)2 | 10 | −6.02 |
| Ni(NO3)2·6H2O | 10 | +4.20 |
| NiCl2 | 10 | +15.96 |
| CuCl2·2H2O | 10 | −42.72 |
| Fe(NO3)3·9H2O | 10 | −3.12 |
| Humic acid | 0.1 | −21.24 |
| Humic acid | 1 | −46.90 |
| Humic acid | 10 | −54.87 |
Significant depressive interferences were observed in the presence of Na2CO3, CoCl2·6H2O, CuCl2·2H2O and humic acid. Little effect was caused by KNO3, Mg(NO3)2·6H2O, CrCl3·6H2O, Cr(NO3)3, K2CrO4, MnCl2·4H2O, Mn(NO3)2·4H2O, Zn, Cd(NO3)2·4H2O, Pb(NO3)2, Ni(NO3)2·6H2O and Fe(NO3)3·9H2O, whereas an enhancement effect was observed for NaCl, KCl, MgCl2·6H2O, CaCO3, CaCl2 and NiCl2.
The interference effect owing to HCl and HNO3 was examined in the range 0–5% v/v (Fig. 6). Results showed that whereas no interference effect was observed for HNO3, the absorbance signal dropped to almost 0 with HCl. HCl caused an interference effect at a concentration as low as 0.1% v/v. This could be due to the stabilization of Hg(II) in the form of chloride complexes that would prevent Hg(II) photoreduction.
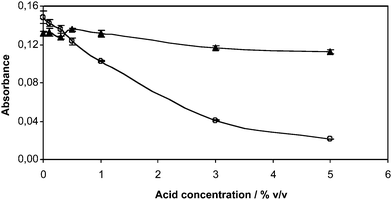 | ||
| Fig. 6 Interference effect of HCl (○) and HNO3 (▲) on the photogeneration of Hg vapour. | ||
UV-photo-induced cold vapor generation from several Hg species
In the UV-photo-induced CVG, both inorganic Hg and organomercury species can be reduced to elemental mercury. It was checked that standard solutions (50 μg L−1 as Hg) of thiomersal, methyl-, ethyl- and phenyl-mercury chlorides, yielded a similar absorbance as compared to a Hg(II) solution. A recovery study showed that complete Hg recovery could be obtained for a variety of Hg species (Table 2). This is a noteworthy finding, since analytical signals for Hg using conventional reducing systems in the cold vapour technique typically depend on the Hg species.| Hg species | Hg spiked/μg L−1 (as Hg) | Hg recovered/μg L−1 (as Hg) | Recovery (%) |
|---|---|---|---|
| MeHg+ | 50 | 48.5 | 97 |
| Thiomersal | 50 | 51.2 | 102 |
| EtHg+ | 50 | 50.9 | 102 |
| PhHg+ | 50 | 48.9 | 98 |
Analytical figures of merits
Under optimal conditions, the calibration function was linear at least up to 100 μg L−1 Hg(II) (r = 0.999). The repeatability expressed as relative standard deviation for N = 15 measurements of a 50 μg L−1 Hg(II) solution was 2.7%. The limit of detection (LOD) (calculated as 3 σ) with stopped-flow was 0.3 μg L−1 of Hg. When no stopped-flow was used, an LOD of about 1.8 μg L−1 of Hg was obtained. The latter LOD is similar to that reported elsewhere for the determination of mercury by photo-CVG-AAS with acetic acid using flow injection without stopped-flow.13 The LOD reported here with our FI/SF-photo-CVG-AAS system is enough to meet the requirements of regulations, where maximum limits in drinking water are 1–2 μg L−1 Hg. The achievement of lower LODs would require preconcentration, e.g., amalgamation, or the use of more sensitive techniques such as atomic fluorescence spectrometry.Application to Hg determination in natural waters
The developed method was applied to the analysis of seven water samples: drinking water, spring water, river water (Tea River, Ponteareas), mineral water (Fontecelta), dam water (Zamáns), river water (Zamáns) and CRM NWTM-27.2 (Ontario Lake) (without Hg). All samples, except the mineral water and CRM, were collected in the north-west of Spain. Blanks were run together with the samples.Analytical results for the water samples are shown in Table 3. The concentration of mercury was below the LOD in all cases. Therefore, spiking experiments were performed in order to test the usefulness of the proposed method. Results showed good recoveries, in the range of 93–103%. The low recovery found in the water from the Tea River could be due to the presence of natural organic matter. This interference effect was overcome following calibration by the standard addition method, which yielded a Hg recovery of 98% for that sample.
| Sample | Hg(II) spiked/μg L−1 | Hg recovered/μg L−1 | Recovery (%) |
|---|---|---|---|
| a Each result represents the mean of three measurements. | |||
| Drinking water | 50 | 50.5 | 101.6 |
| Spring water | 50 | 50.4 | 100.9 |
| Tea River | 50 | 46.3 | 92.7 |
| Mineral water (Fontecelta) | 50 | 50.8 | 101.6 |
| Dam water (Zamáns) | 50 | 47.6 | 95.2 |
| River water (Zamáns) | 50 | 48.1 | 96.2 |
| CRM NWTM-27.2 | 50 | 51.4 | 102.9 |
Conclusions
Photoassisted reduction of Hg species to yield Hg (0) can be performed in a FI/SF-photo-CVG-AAS system without the need for a chemical reducing agent and a mineral acid at high concentration. Stopped-flow operation overcomes the occurrence of kinetically-slow photoreduction reactions, thus improving the LOD when flow-injection systems are employed. This approach allows larger doses of UV radiation to be provided to the sample zone in the flow-through photoreactor. Remarkably, the same photoreduction efficiency is observed regardless of the Hg species injected in the FI/SF manifold.This approach can be applied as a simple, low-cost and ecofriendly technique for determination of Hg(II) in natural freshwater samples in the presence of a low molecular weight organic acid. Best results were obtained using acetic acid in comparison with oxalic, citric and ethylenediaminetetraacetic acids.
Acknowledgements
Financial support from the Spanish Ministry of Science and Innovation (projects CTQ2006-04111/BQU and CTQ2009-06956/BQU) and the Vigo University (Contract for Reference Research Groups 09VIA08) are gratefully acknowledged.References
- L. I. Sweet and J. T. Zelikoff, J. Toxicol. Environ. Health, Part B, 2001, 4, 161–205 CAS.
- U.S. Environmental Protection Agency. Mercury Sources and Regulations. Draft, November 1 (1999),http://www.epa.gov/bns/mercury Search PubMed.
- K. Wang, K. D. Kim, D. D. Dionysiou, G. A. Sorial and D. Timberlake, Environ. Pollut., 2004, 131, 323–336 CrossRef CAS.
- GPO website, page for 40 CFR part 141, http://a257.g.akamaitech.net/7/257/2422/14mar20010800/edocket.access.gpo.gov/cfr_2002/julqtr/40cfr141.62.htm.
- EU (European Communities) Commision Directive 2003/40/EC of May 16 (2003) on natural mineral waters.
- M. Cullen, in Atomic Spectroscopy in Elemental Analysis, Blackwell Publishing, Oxford, UK, 2004, pp. 241–254 Search PubMed.
- R. Sturgeon, Anal. Bioanal. Chem., 2007, 388, 733–734 CrossRef CAS.
- Y. He, X. Hou, C. Zheng and R. E. Sturgeon, Anal. Bioanal. Chem., 2007, 388, 769–774 CrossRef CAS.
- X. Guo, R. E. Sturgeon, Z. Mester and G. J. Gardner, Anal. Chem., 2004, 76, 2401–2405 CrossRef CAS.
- C. Zheng, Y. Li, Y. He, Q. Ma and X. Hou, J. Anal. At. Spectrom., 2005, 20, 746–750 RSC.
- M. A. Viera, A. S. Ribeiro, A. J. Curtius and R. E. Sturgeon, Anal. Bioanal. Chem., 2007, 388, 837–847 CrossRef CAS.
- J. T. Madden and N. Fitzgerald, Spectrochim. Acta, Part B, 2009, 64, 925–927 CrossRef.
- R. F. Bendl, J. T. Madden, A. L. Regan and N. Fitzgerald, Talanta, 2006, 68, 1366–1370 CrossRef CAS.
- A. López-Rouco, E. Stanisz, H. Matusiewicz and C. Bendicho, J. Anal. At. Spectrom., 2008, 23, 1026–1029 RSC.
- L. Wu, C. Zheng, Q. Ma, C. Hu and X. Hou, Appl. Spectrosc. Rev., 2007, 42, 79–102 CrossRef CAS.
- H. M. Li, Y. Zhang, C. B. Zheng, L. Wu, Y. Lv and X. Hou, Anal. Sci., 2006, 22, 1361–1365 CrossRef CAS.
- Y. Yin, J. Liang, L. Yang and Q. Wang, J. Anal. At. Spectrom., 2007, 22, 330–334 RSC.
- Y. Yin, J. Qiu, L. Yang and Q. Wang, Anal. Bioanal. Chem., 2007, 388, 831–836 CrossRef CAS.
- Y. Li, C. Zheng, Q. Ma, L. Wu, C. Hu, X. Hou and J. Anal, J. Anal. At. Spectrom., 2006, 21, 82–85 RSC.
- A. Han, C. Zheng, J. Wang, G. Cheng, Y. Lv and X. Hou, Anal. Bioanal. Chem., 2007, 388, 825–830 CrossRef CAS.
- M. García, R. Figueroa, I. Lavilla and C. Bendicho, J. Anal. At. Spectrom., 2006, 21, 582–587 RSC.
| This journal is © The Royal Society of Chemistry 2010 |