A homogeneous time-resolved fluoroimmunoassay (TR-FIA) using antibody mediated luminescence quenching
Frank
Sellrie
*a,
Michael
Beck†
b,
Niko
Hildebrandt
c and
Burkhard
Micheel
d
aUP Transfer GmbH—Hybrotec, c/o Fraunhofer Institute for Biomedical Engineering, Am Mühlenberg 13, D-14476, Potsdam-Golm, Germany. E-mail: frank.sellrie@up-transfer.de
bPhysical Chemistry, University of Potsdam, Karl-Liebknecht-Str. 24-25, D-14476, Potsdam-Golm, Germany
cNanoPolyPhotonics, Fraunhofer Institute for Applied Polymer Research, Geiselbergstr. 69, D-14476, Potsdam-Golm, Germany
dDepartment of Biotechnology, Institute of Biochemistry and Biology, University of Potsdam, Karl-Liebknecht-Str. 24-25, D-14476, Potsdam-Golm, Germany
First published on 22nd July 2010
Abstract
The determination of low-molecular weight substances (haptens) is demonstrated with a homogeneous time-resolved immunoassay using antibody-induced luminescence quenching. Our novel assay technology uses the newly developed monoclonal antibody (G24-BA9) to quench the luminescence of europium trisbipyridine (EuTBP). We performed a competitive biotin immunoassay including an EuTBP–biotin conjugate, the anti-EuTBP antibody G24-BA9 and streptavidin as assay components. Steric hindrance allows only the binding of either G24-BA9 (to the EuTBP moiety) or streptavidin (to the biotin moiety) to the EuTBP–biotin conjugate. Addition of the analyte biotin resulted in the binding of streptavidin to biotin and a concomitant preferred binding of G24-BA9 to EuTBP–biotin. Since G24-BA9 quenches the luminescence of EuTBP within the conjugate, the luminescence signal could be used to indicate and quantify the presence of free biotin in the system. All experiments were carried out in solution in the presence of 5% serum demonstrating the possibility of using our novel assay for a very fast determination of low molecular weight substances in biological fluids.
Introduction
Homogeneous immunoassays allow the fast determination of analytes in a single test step without tedious and time-consuming separation procedures. Nonetheless, the total number of homogeneous immunoassays commercially available is small compared to heterogeneous systems.Fluorescence offers several possibilities to establish sensitive homogeneous immunoassays.1 Polarisation of light emitted by fluorescent dyes,2 Förster resonance energy transfer (FRET) between two different fluorescent dyes3–5 and fluorescence quenching by antibodies6 have been used for such assays.
A typical direct fluorescence quenching immunoassay uses an anti-analyte antibody, an analyte–fluorophore conjugate and the competing free analyte.7,8 Since in the conjugate the fluorophore and the analyte are in very close proximity binding of the anti-analyte antibody results in quenching of the fluorescence of the fluorophore. Another approach uses antibody binding for quenching FRET within analyte–fluorophore thus increasing the fluorophore fluorescence intensity.9,10 Another type of fluorescence quenching immunoassay designated indirect quenching fluoroimmunoassay or fluorescence protection immunoassay implies the basic principle of the experiments presented here.11,12 This type of assay uses an analyte–fluorophore conjugate together with two antibodies, one reactive to the analyte and the other binding and quenching the fluorophore. Due to steric hindrance, simultaneous binding of both antibodies to the conjugate is not possible so that the addition of free analyte changes the equilibrium of the system and hence also changes the fluorescence of the fluorophore in the conjugate.
We applied such an assay type for the determination of the herbicide diuron using fluorescein as fluorophore and the anti-fluorescein antibody B13-DE1 as quencher.13 Fluorescein is, however, not suitable as indicator for the determination of substances in biological fluids as its fluorescence is quenched by high protein concentration and is also spoiled by autofluorescence background signals.
Lanthanide complexes of europium (Eu3+) and terbium (Tb3+) ions are advantageous fluorescent markers for immunoassays since their long luminescence decay times (up to milliseconds) allow a time-gated fluorescence measurement and therefore an efficient discrimination from the short-lived autofluorescence background.14,15
Herein, we describe the determination of biotin in an indirect time-resolved fluorescence quenching immunoassay using a europium trisbipyridine (EuTBP) cryptate16–18 conjugated to biotin as indicator, streptavidin as biotin binder and an anti-EuTBP monoclonal antibody (G24-BA9) as quencher. The assay is very fast and sensitive and avoids the problems occurring when fluorescein is used as fluorescent moiety.
Experimental
EuTBP–biotin conjugate
Biotin–NHS (biotin N-succinimidyl ester; Fluka, Buchs, Switzerland) was conjugated to the europium diamino cryptate (EuTBP-NH2) in a molar ratio of 3 to 1. EuTBP-NH2 (europium(III) trisbipyridine cryptate-4,4′-bis(2-aminoethyl-aminocarbonyle) penta-trifluoroacetate salt) was purchased from Cisbio, Bagnols sur Cèze, France. An excess of the biotin derivative was chosen to ensure a conjugation of both amino groups.1 µl biotin–NHS containing 7 mg ml−1 was added to 1 µl containing 1 mg ml−1 EuTBP-NH2. The reaction was carried out in carbonate/bicarbonate buffer (50 mM, pH 9) at room temperature for 3 hours.
The resulting EuTBP–biotin conjugate (molecular weight presumably 1380 Da if both NH2 groups were conjugated) is presented in Fig. 1. It was used in all experiments without further purification.
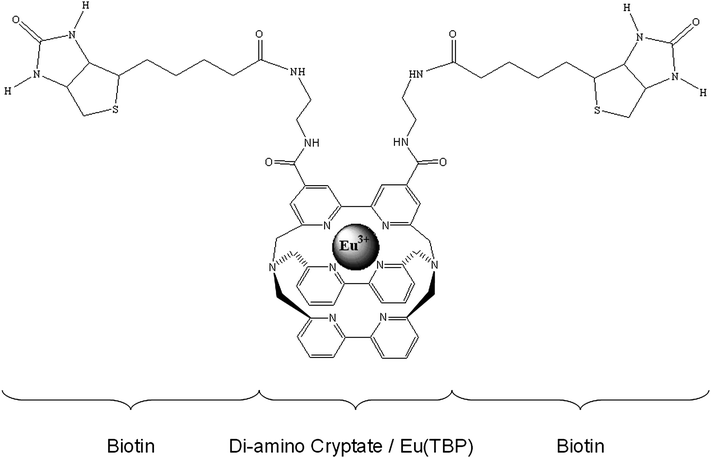
Generation of a EuTBP luminescence quenching monoclonal antibody
A monoclonal EuTBP-specific antibody was generated by hybridoma technology.19 For this purpose a female Balb/c mouse was immunized three times with an EuTBP–streptavidin conjugate. Immunization started with 25 µg conjugate using Freund's complete adjuvant. Booster immunizations were carried out six and eight weeks after the first immunization using 10 µg conjugate without adjuvant. Four days after final booster immunization electrofusion of spleen cells with myeloma cells (Sp2/0, ATCC CRL-1581) in the presence of polyethylene glycol 8000 was performed as described.20 Selected hybrids were cultivated in RPMI 1640 medium (containing 10% FCS, 2 mM glutamine and 50 mM β-mercaptoethanol) and subcloned by limiting dilution on mouse peritoneal feeder cells. Culture supernatants of clones and subclones were tested in an enzyme immunoassay for antigen binding. An EuTBP–bovine serum albumin (BSA) conjugate was adsorbed to the solid phase. The class and subclass of monoclonal antibodies were determined as described.20 Purification of antibody from culture supernatant was performed using protein A affinity chromatography.21 Resulting antibodies were tested for quenching the luminescence of free and protein bound EuTBP.Equipment
A modified KRYPTOR system (Cezanne, Nîmes, France) was applied for time-resolved integrated single photon counting at one photomultiplier (PMT) channel (620 nm for Eu, filter based wavelength separation) with 50 µs delay, 400 µs gate using fiber-coupled laser excitation (N2-laser, 337.1 nm excitation wavelength). The samples were measured in a multi-well plate with 300 wells of 150 µl sample volume each.Antibody-induced Eu(TBP)–biotin luminescence quenching
Mixtures of diluted EuTBP–biotin conjugate (dilution 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :300000 of described conjugate) with diluted purified anti-EuTBP antibodies were checked for their luminescence in comparison to samples without antibodies. The final antibody concentration was varied from 20 to 0.35 µg ml−1. Experiments were carried out in phosphate-buffered saline (PBS) containing 5% newborn calf serum (final volume 150 µl per sample).
:300000 of described conjugate) with diluted purified anti-EuTBP antibodies were checked for their luminescence in comparison to samples without antibodies. The final antibody concentration was varied from 20 to 0.35 µg ml−1. Experiments were carried out in phosphate-buffered saline (PBS) containing 5% newborn calf serum (final volume 150 µl per sample).
Luminescence quenching assay for the determination of biotin
The assay for the determination of biotin was performed on the basis of the quenching assay described in the previous section. Antibody G24-BA9 in a final concentration of 2.5 µg ml−1 was mixed with the EuTBP–biotin conjugate and the analyte biotin (in final concentrations of 7 to 42 µg ml−1). Streptavidin (final concentration 2.5 µg ml−1) was added in the last step. All dilutions were performed in the same buffer as above containing newborn calf serum. Luminescence measurements were carried out immediately after mixing.Results and discussion
A monoclonal antibody binding EuTBP was generated by hybridoma technology using an EuTBP–streptavidin conjugate for immunization. The serum of the immunized mouse was quenching the luminescence of EuTBP and the spleen-cells of the mouse were, therefore, chosen for fusion (data not shown). Only one monoclonal antibody of IgG1 subclass designated G24-BA9 could be isolated in the course of the experiments described here. Since this antibody did not only bind EuTBP conjugates but also quenched the luminescence of free EuTBP all experiments were performed using this antibody in purified form. The luminescence of the EuTBP–biotin conjugate was quenched in a similar way as the free hapten. Luminescence quenching of EuTBP–biotin conjugate is presented in Fig. 2.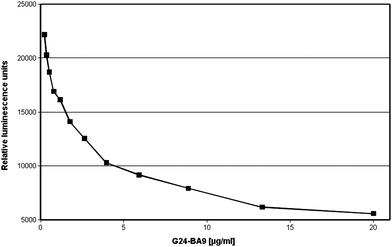 | ||
| Fig. 2 Luminescence quenching of EuTBP–biotin induced by the binding of the monoclonal EuTBP-binding antibody G24-BA9 to the conjugate. The assay mixture consisted of EuTBP–biotin conjugate (final dilution 1:300000) and the antibody G24-BA9 (standard dilutions of 0.35 to 20 µg ml−1) in a final volume of 150 µl phosphate-buffered saline (PBS) containing 5% newborn calf serum. | ||
A homogeneous indirect luminescence quenching immunoassay for the determination of biotin could be developed on the basis of this effect. The assay system consisted of an EuTBP–biotin conjugate (Fig. 1), the quenching antibody G24-BA9 and the biotin binder streptavidin. The indicator molecule EuTBP–biotin can be bound either by antibody G24-BA9 or by streptavidin. Due to steric hindrance, simultaneous binding of both molecules to the conjugate is not possible. Free biotin in the mixture should result in the binding by streptavidin so that more anti-EuTBP antibody can bind to the conjugate. Since G24-BA9 quenches the luminescence of EuTBP in the conjugate a diminished luminescence signal can be used as an indicator for the presence of the analyte biotin. The interaction between the assay components and the analyte and the resulting luminescence are schematically shown in Fig. 3.
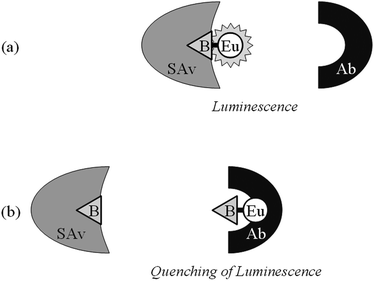 | ||
| Fig. 3 Schematic presentation of the homogeneous luminescence quenching immunoassay. A homogeneous immunoassay was established for the determination of the hapten analyte biotin. (a) The assay system contains a conjugate in which the analyte biotin (B) is chemically linked to the luminophore EuTBP (Eu) and two different binding molecules, one specific for the analyte (streptavidin—SAv) and the other for the luminophore (antibody G24-BA9—Ab). The proximity of B and Eu in the conjugate permits the binding of only one binding molecule at a time. (b) Since Ab quenches the luminescence of Eu, the steric hindrance can be utilized to demonstrate free B added to the system. Addition of free B reduces the amount of conjugate-bound SAv and increases the amount of conjugate-bound Ab, which induces the luminescence quenching. The decreasing luminescence intensity is therefore a quantitative indicator of free analyte present in the test sample. | ||
The assay system was successfully applied to the determination of biotin. Increasing analyte concentrations resulted in decreasing luminescence values as expected. A concentration of 18.1 ng ml−1 biotin led to a 50% reduction in the luminescence signal in our model experiment (Fig. 4).
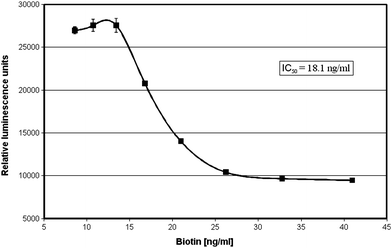 | ||
| Fig. 4 Application of the homogeneous luminescence quenching immunoassay for the determination of the vitamin biotin. The assay mixture consisted of an EuTBP–biotin conjugate (final dilution 1:300000), the monoclonal antibody G24-BA9 (final concentration 20 µg ml−1) and streptavidin (final concentration 2.5 µg ml−1), as well as the test samples to be checked for the presence of biotin (standard dilutions of 7–42 ng ml−1) in a final volume of 150 µl phosphate-buffered saline (PBS) containing 5% newborn calf serum. A concentration of 18.1 ng ml−1 biotin leads to a 50% reduction in luminescence. | ||
The main advantage of this assay is the very short evaluation time since it does not depend on any long-time incubation or separation steps. The assay was carried out in the presence of 5% newborn calf serum which had obviously no negative effects on the results. Higher protein concentrations are generally unfavourable for most fluorescence assay applications.13 A successful application of our novel assay principle was possible because of the utilization of time-resolved luminescence detection which easily discriminates the assay signals from short-lived autofluorescence background signals.
The assay should allow the determination of biotin (vitamin H) in different tissue and food samples.22 For the determination and quantification of biotin in serum the detection limit of the assay has to be improved by a factor of 10 to detect concentrations in a range of 0.2 to 1 ng ml−1.23 This problem can possibly be solved by purifying the conjugate which presumably still contains free unconjugated biotin which may disturb the assay and impairs the sensitivity.
One other problem affecting the assay sensitivity might be related to the fact that streptavidin is capable of binding four biotin molecules. This could in principle lower the sensitivity by a factor of four. It is otherwise probably responsible for a cross-linking between streptavidin and EuTBP–biotin (as one conjugate molecule consists of two biotin molecules). The concentrations used in the experiments are too low to observe any visible effects like precipitation or turbidity. A cross-linking would increase the steric hindrance of antibody binding and therefore even improve the assay sensitivity. Experiments confirming these assumptions were not carried out.
After reaching a better sensitivity this assay could be the most favourable test for the determination of biotin especially because of its simple sample handling and very fast evaluation time compared to the biotin determinations applied so far.24
Our homogeneous assay should also be applicable for the determination of other low molecular weight substances. For this purpose monoclonal antibodies have to be selected binding to the corresponding analyte and competing with the anti-EuTBP antibody for binding to the analyte–EuTBP conjugate. The big advantage is that only one new antibody needs to be selected because the anti-EuTBP antibody can be used for all different assays.
Due to the necessity of steric hindrance it is much more difficult to apply this assay type to the determination of high-molecular weight substances. However, Nargessi et al.11 and Zuk et al.12 demonstrated that steric hindrance of antibodies in binding to the surface of fluorophore-labeled proteins can be used in some cases to generate homogeneous immunoassays. Another approach could be the use of peptides in the luminophore conjugates representing linear or mimotope25,26 epitopes of the corresponding proteins.
The selection of the necessary antibodies requires, however, considerable efforts. Investigations for evaluating the feasibility of these approaches towards the determination of proteins with our assay system are in progress.
Conclusion
A fast homogeneous luminescence quenching immunoassay was developed on the basis of time-resolved luminescence detection. The assay utilizes luminescence quenching of EuTBP (europium trisbipyridine cryptate) by the monoclonal antibody G24-BA9 and the steric hindrance-based binding competition (for the EuTBP–analyte conjugate) between G24-BA9 (to the EuTBP moiety) and an analyte binding molecule (to the analyte moiety). Using time-resolved luminescence measurements allows the determination of analytes in the presence of higher protein concentrations, as e.g. in biological fluids because the assays autofluorescence can be efficiently suppressed. This immunoassay can therefore be used to determine a wide variety of substances of medical and environmental interest. We expect our novel assay to be easily integrated in a nanoscale microfluidic application.Acknowledgements
We thank Dr K. Messerschmidt (University of Potsdam, Biotechnology) for technical assistance concerning cell fusion. We gratefully acknowledge the support of J. A. Schenk.References
- E. Soini and I. Hemmilä, Clin. Chem. (Washington, DC, U. S.), 1979, 25, 353 CAS.
- D. S. Smith and S. A. Eremin, Anal. Bioanal. Chem., 2008, 391, 1499 CrossRef CAS.
- K. Blomberg, P. Hurskainen and I. Hemmilä, Clin. Chem. (Washington, DC, U. S.), 1999, 45, 855 CAS.
- S. A. Kane, C. A. Fleener, Y. S. Zhang, L. J. Davis, A. L. Musselman and P. S. Huang, Anal. Biochem., 2000, 278, 29 CrossRef CAS.
- I. Hemmilä and S. Webb, Drug Discovery Today, 1997, 9, 373 CrossRef.
- E. W. Voss, Jr, J. Mol. Recognit., 1993, 6, 51 CrossRef CAS.
- E. J. Shaw, R. A. Watson, J. Landon and D. S. Smith, J. Clin. Pathol., 1977, 30, 526 CrossRef CAS.
- I. Hemmilä, O. Malminen, H. Mikola and T. Lovgren, Clin. Chem. (Washington, DC, U. S.), 1988, 34, 2320 CAS.
- C. Tan, N. Gajovic-Eichelmann, W. F. M. Stöcklein, R. Polzius and F. F. Bier, Anal. Chim. Acta, 2009, 658, 187.
- C. Tan, N. Gajovic-Eichelmann, R. Polzius, N. Hildebrandt and F. F. Bier, Anal. Bioanal. Chem., 2010 Search PubMed , submitted.
- R. D. Nargessi, R. J. Landon and D. S. Smith, J. Immunol. Methods, 1979, 26, 307 CrossRef CAS.
- R. F. Zuk, G. L. Rowley and E. F. Ullman, Clin. Chem. (Washington, DC, U. S.), 1979, 25, 1554 CAS.
- F. Sellrie, A. Warsinke and B. Micheel, Anal. Bioanal. Chem., 2006, 386, 206 CrossRef CAS.
- I. Hemmilä, J. Biomol. Screening, 1999, 4, 303 CrossRef CAS.
- I. Hemmilä and V. Laitala, J. Fluoresc., 2005, 15, 529 CrossRef CAS.
- G. Mathis, Clin. Chem., 1995, 41, 1391 CAS.
- H. Bazin, M. Preaudat, E. Trinquet and G. Mathis, Spectrochim. Acta, Part A, 2001, 57, 2197 CrossRef CAS.
- H. Bazin, E. Trinquet and G. Mathis, J. Biotechnol., 2002, 82, 233 CAS.
- G. Köhler and C. Milstein, Nature, 1975, 256, 495.
- J. A. Schenk, F. Matyssek and B. Micheel, In Vivo, 2004, 18, 649 Search PubMed.
- R. Lindmark, K. Thorén-Tolling and J. Sjöquist, J. Immunol. Methods, 1983, 62, 1 CrossRef CAS.
- C. G. Staggs, W. M. Sealey, B. J. McCabe, A. M. Teague and D. M. Mock, J. Food Compos. Anal., 2004, 17, 767 CrossRef CAS.
- S. I. Hansen and J. Holm, Clin. Chem. (Washington, DC, U. S.), 1989, 35, 1721 CAS.
- E. Livaniou, D. Costopopoulou, I. Vassiliadou, L. Leondiadis, J. O. Nyalala, D. S. Ithakissios and G. P. Evangelatos, J. Chromatogr., A, 2000, 881, 331 CrossRef CAS.
- V. Böttger, L. Peters and B. Micheel, J. Mol. Recognit., 1999, 12, 191 CrossRef CAS.
- F. Sellrie, J. A. Schenk, O. Behrsing, V. Böttger and B. Micheel, J. Immunol. Methods, 2002, 261, 141 CrossRef CAS.
Footnote |
| † Current address: Einstein-Gymnasium, Haydnstr. 3, D-77694 Kehl, Germany |
| This journal is © The Royal Society of Chemistry 2010 |