A simple kinetic spectrophotometric method for simultaneous determination of tetracyclines by use of chemometrics
Yongnian
Ni
*ab,
Na
Deng
b and
Serge
Kokot
c
aKey Laboratory of Food Science and Technology, Nanchang University, Nanchang, jiangxi 330047, China
bDepartment of Chemistry, Nanchang University, Nanchang, Jiangxi 330047, China. E-mail: ynni@ncu.edu.cn; Fax: +86 791 3969500; Tel: +86 791 3969500
cChemistry, Faculty of Science and Technology, Queensland University of Technology, Brisbane, Queensland 4001, Australia
First published on 23rd July 2010
Abstract
A new kinetic spectrophotometric method for simultaneous determination of chlortetracycline, tetracycline and oxytetracycline in real samples has been researched and developed. The method was based on the kinetic reaction involving the oxidation of the drugs by alkaline potassium permanganate at 40 °C. After the addition of these drugs, an increasing absorbance at 526 nm and a decreasing absorbance at 608 nm caused by manganate and permanganate, respectively, were observed. The three-way kinetic-spectral data in the 325–725 nm range from 0 to 360 s were collected and applied to build chemometrics models. The plot of absorbance versus concentration was rectilinear over the 0.05–0.75 µg mL−1 range for chlortetracycline and tetracycline, and 0.02–0.34 µg mL−1 for oxytetracycline, with the limit of detection of 0.013, 0.015 and 0.011 µg mL−1, respectively. The experimental conditions were carefully investigated to optimize the development and stability of the color products (potassium manganate). The proposed method was successfully applied to the simultaneous determination of tetracyclines (TCs) in feed and muscle samples, and the results showed satisfactory agreement with those from of the HPLC method.
Introduction
Tetracyclines (TCs) have a broad range of activity against gram-positive and gram-negative bacteria,1,2 and have been commonly used in veterinary medicine for disease prevention and treatment. The widespread use of tetracyclines could lead to the existence of residues in animal-producing foods. The basic structure of tetracyclines consists of a hydronaphthacene framework containing four fused rings.3 Because of the possible toxicity of their drug residues in food and the emergence of drug-resistant pathogenic organisms,3,4 the administration of tetracyclines has been the focus of attention recently. The U.S. Food and Drug Administration's Center for Veterinary Medicine (FDA-CVM) has set tolerances for the sum of residues of the tetracyclines including chlortetracycline, oxytetracycline and tetracycline in muscle of 100 µg kg−1 and in liver of 300 µg kg−1,5 while in European Union (EU) the maximum residues limits (MRLs) have been established as 600 µg kg−1 in liver and 200 µg kg−1 in muscle for the three tetracyclines.6Several popular methods for the determination of tetracyclines were reported, including HPLC,7,8 thin layer chromatography,9 capillary electrophoresis (CE),10 FIA coupled to CE,11,12 microbiological assay,13 immunoassay,14 and stopped flow mixing.15 Besides methods involving a pre-separation procedure such as chromatography and CE, few other approaches have been developed for the simultaneous determination of tetracyclines. As an alternative to those methods, spectrofluorometric and luminescent techniques combined with multivariate calibration have been proposed16,17 for individual quantification of tetracyclines. Recently, a method using photochemically induced fluorescence signals combined with both first- and second-order multivariate calibration have been reported to quantify a mixture of three tetracyclines in surface water samples with satisfactory results.18,19
Differential kinetic methods for which two or more species react with a common reagent could be used to resolve mixtures without prior separation. The most severe shortcoming of classical computational techniques based on kinetic data is that they require a prior knowledge of the system studied, namely, of the order and rate constant for each analyte involved.20 Chemometrics methods such as principal component regression (PCR),21 partial least squares (PLS),22 artificial neural networks (ANN),23 as well as some three-way methods, N-way partial least squares (NPLS), unfolded partial least squares (UPLS) and unfolded principal component regression (UPCR) have been successfully applied to the multicomponent kinetic determination to overcome above drawbacks. Any non-linearities may be reduced or overcome by increasing the number of principle components in the model, which then can be used for various complex systems without any prior knowledge of all the system's components present. Thus, this approach is particularly applicable to kinetic systems that do not always behave in an ideal or expected manner.24
In this paper, a rapid, simple and sensitive differential kinetic method was researched and developed for the determination of chlortetracycline, tetracycline and oxytetracycline in feed and muscle samples, and it was based on the redox reaction between the tetracyclines and alkaline potassium permanganate. Calibration models of PCR, PLS, RBF-ANN, NPLS, UPLS and UPCR were established from the kinetic data of the standard analyte mixtures, and then the chemometrics models were verified, compared, and applied to predict tetracyclines in real samples.
Theoretical background
Methodology
Consider that there are s analytes, Mj (j = 1, 2, …, s), which react with a common absorbing reagent, R (in this work, the potassium permanganate), to give a common absorbing product, P (potassium manganate), and i different non-absorbing products, Qj, according to: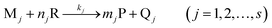 | (1) |
If the concentration of R is much larger than those of Mj, i.e.cR ≫ cMj, the reaction can be considered as a pseudo-first-order kinetics for the analytes.25
Thus, its rate equation for a certain analyte, Mj, can be represented as:
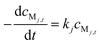 | (2) |
| cMj,t = cMj,0 exp(−kjt) | (3) |
According to eqn (1), the concentration of R or P at time t, cRj,t and cPj,t respectively, can be represented as follows:
| cRj,t = cRj,0 − njcMj,0(1 − exp(−kjt)) | (4) |
| cPj,t = mjcMj,0(1 − exp(−kjt)) | (5) |
Assume that there is an overlap between the spectra of R and P in the wavelength range studied and their absorbances obey the Beer–Lambert law at any wavelength, λi, the absorbance, AMj,λ,t, of Mj at wavelength, λ, and time, t, can be written as:
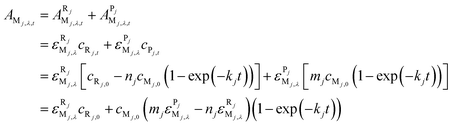 | (6) |
Let cRj,0 = acMj,0 (a is the proportional coefficient here), and then eqn (6) can be further simplified to:
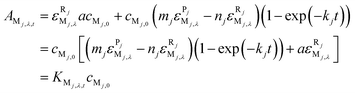 | (7) |
If s analytes participate in this reaction, and the absorbance given by these analytes are additive, eqn (7) can be extended as the total absorbance of s analytes:
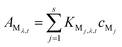 | (8) |
If m standard samples are prepared, the absorbance data of kinetic systems being monitored at p wavelengths and total k time points, eqn (8) can then be expressed in matrix form as follows:
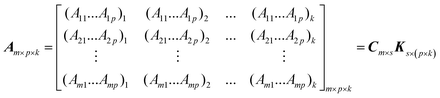 | (9) |
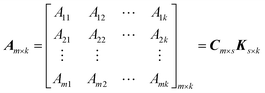
At a given wavelength point, eqn (9) can be reduced to the kinetic form:
 | (10) |
Chemometrics for two-way data
PCR and PLS are multivariate regression techniques, which have been successfully applied to many analytical systems.26,27 PCR and PLS need a calibration step where the models for the response spectra and the analyte concentration are deduced from a set of standards, followed by a prediction step, in which the concentrations of unknowns are estimated from the sample spectrum. Both of these two methods involve spectral decomposition. The PCR decomposition is based entirely on spectral variations without regard for analyte concentrations, and the PCR decomposition is significantly influenced by variations, which have no relevance to the analyte concentrations, while for PLS, the spectral decomposition is weighted to the concentration. Both PCR and PLS are factor analysis based methods and have some major advantages, for example, they do not need a knowledge of all the components in the mixture, and can partially overcome the non-linearities of the complex system by increasing the factor numbers. The choice of these two calibration methods often depends on the particular experimental conditions.28,29Artificial neural networks (ANNs) are powerful chemometrics methods because they do not need any model structure speciation and can process multivariate problems of nonlinear systems. With proper training, ANNs can accurately model the presence of synergistic effects and avoid the potential loss of kinetic data for mixtures resulting from too short induction periods, outliers, small differences in the rate constants, and so on. The principles of ANNs can be found elsewhere.30
Chemometrics for three-way data
The main feature of the NPLS algorithm is that it produces score vectors, which in a trilinear sense have maximum covariance with the unexplained part of the dependent variable.31 In the case of three-way data, the model is given by the following equation: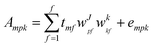 | (11) |
Both the unfolded partial least squares (UPLS) and unfolded principal component regression (UPCR)32–34 consist of the two-way calibration PLS and PCR models when they are applied to three-way data that has been unfolded into vectors. With such an unfolded matrix, usual PLS and PCR models can be built and applied to solve quantitative problems for three-way data.
Experimental
Reagents and solutions
All reagents were of analytical-reagent grade and used without further purification. Aqueous stock solutions of chlortetracycline, tetracycline and oxytetracycline, 100 mg L−1 for each, were prepared by dissolving suitable their crystalline (Sigma, USA) in 10 mL of 0.1 mol L−1 sodium hydroxide and diluted to 100 mL with water. There stock solutions were appropriately diluted for practical use. A stock solution of 1.0 mol L−1 sodium hydroxide was prepared by dissolving 20.0 g of sodium hydroxide in 500 mL of water. A stock solution of 0.01 mol L−1 potassium permanganate was prepared by dissolving 0.395 g potassium permanganate in 250 mL water. Doubly distilled water was used throughout.Instrumentation and software
An Agilent 8453 UV-visible spectrophotometer was used for spectra and kinetic curves acquisition. All measurements were performed in a thermostated cell compartment at 40 ± 0.5 °C by means of a Model ZC-10 (Ningbo Tianheng Instruments Factory, China) temperature control accessory.The high-performance liquid chromatography (HPLC) procedure was carried out by an Agilent 1100 series HPLC-DAD system equipped with a vacuum degasser, quaternary pump, autosampler, injector with a 100 µL loop, an Agilent ZORBAX Eclipse XDB-C18 column (4.6 mm × 250 mm, 5 µm) with an Agilent Zorbax high pressure reliance cartridge guard-column (C18, 12.5 mm × 4.6 mm, 5 µm) and a DAD detector. The optimized experimental parameters were: (A) mobile phase, 20% KH2PO4:H3PO4 buffer (0.2 mol L−1, pH 2.5) and (B) methanol with a gradient program of 45–55% (B, v/v) during 0–20 min; flow rate, 1 mL min−1; temperature, 27 °C; detector wavelength, 244 nm for chlortetracycline, tetracycline and oxytetracycline; retention times, 4.79 min for chlortetracycline, 5.55 min for tetracycline and 8.35 min for oxytetracycline.
General experimental procedure
The reaction was carried out in a 10 mm cell. Taking into account that the total useful volume was 2.50 mL, an appropriate amount of the standard solution of chlortetracycline, tetracycline and oxytetracycline (or their mixture) was pipetted directly into the cell; this was followed by the additions of 0.60 mL of 1.0 mol L−1 NaOH solution and an appropriate volume of water to give a volume of 2.25 mL. The solution was stirred by an auto-controlled micro-stirrer, and left to stand for 2 min in a controlled-temperature (40 °C) cell holder in the spectrophotometer. Subsequently, 0.25 mL of 0.01 mol L−1 potassium permanganate solution was micropipetted to make up the total volume of 2.50 mL. The absorbance of this solution was then measured as soon as possible. The kinetic profiles were blank-corrected automatically in the range of 325 to 725 nm, every 1 nm, at an interval of 2 s, between 0 and 360 s.For HPLC procedure, this reference method is described in detail elsewhere GB/T 5009.116-2003.35 About 10 µL of each TC sample (solution obtained as described in the Section: Preparation of samples) were injected and processed as above. The concentrations of the compounds were evaluated on the basis of the peak area ratios compared to those of the standard solutions.
Preparation of samples
The muscle sample was cut into pieces and blended. Tissue weight aliquots (5.0 g) in triplicate were placed into 50 mL Erlenmeyer flasks (with a screw cap), each containing 25.0 mL 5% perchloric acid; these three samples were spiked in order with 0.25, 0.5 and 1.0 mL of each TC (100 µg mL−1 stock solutions), and then shaken for 1.5 h. For each flask, this mixture was then transferred to a 10 mL centrifuge tube, and centrifuged at 4000 rpm for 10 min. The clear portion of the mixture in the tube was filtered (qualitative filter paper-standard grade) and transferred to a 10 mL flask, then diluted to the mark with methanol. These solutions were then analysed as described (Section: General experimental procedure).Similarly, weight aliquots (5.0 g) of finely ground swine feed, in triplicate, and 50 mL of freshly prepared ethyl acetate extraction solution was added. The mixture was shaken vigorously for 20 min and then centrifuged at 3000 rpm for 10 min. The supernatant was collected, and the residue was extracted repeatedly with the same procedure. Finally, the supernatant was combined and evaporated to dryness, and then made up to 50 mL with methanol.
Results and discussion
Reaction kinetics of tetracyclines
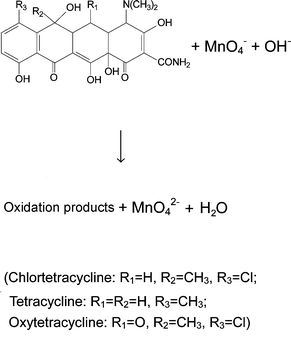
The UV-vis spectra of chlortetracycline, tetracycline and oxytetracycline overlap significantly (figures not shown), and therefore, it is difficult to determine them simultaneously, directly from such spectra. The molecular structures of the tetracyclines show that they all have reductive properties, and thus can react with alkaline potassium permanganate. The incorporation of the redox reactions of these compounds facilitated the kinetic determination, and enabled the development of a differential kinetic spectrophotometric procedure as presented below.In this paper, the reaction kinetics based on the reaction of potassium permanganate with tetracyclines in alkaline medium has been investigated, and the reaction mechanism can be explained as follows:
 | (12) |
Absorbance spectra of the coloured product were measured over the range of 325–725 nm at a reaction time of 360 s (Fig. 1). Their peaks overlapped strongly at 526 and 608 nm, which complicates the analysis of the spectral output from their mixtures. Kinetic curves of chlortetracycline, tetracycline and oxytetracycline were plotted by taking the readings at the absorption peak maxima (526 and 608 nm) in Fig. 2. It is clear from these plots that the reaction rate of chlortetracycline is lower than that of tetracycline and oxytetracycline. Thus, differential kinetic rates may be useful for resolving a mixture of tetracyclines with the use of chemometrics methods. The rate constants for chlortetracycline, tetracycline and oxytetracycline were estimated by fitting the kinetic data from several given single component samples to equation, A = a0 − a1exp(−kt) by a suitable regression method36 are 0.0062, 0.0086 and 0.0112 s−1 at 526 nm and 0.0041, 0.0072 and 0.0098 s−1 at 608 nm for chlortetracycline, tetracycline and oxytetracycline, respectively. There are only small differences between the reaction rates of chlortetracycline, tetracycline and oxytetracycline. Therefore, two-way chemometrics methods, such as PCR, PLS and RBF-ANN, can be applied for analysis of the three tetracyclines.
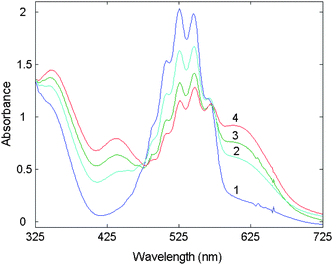 | ||
| Fig. 1 Absorption spectra at t = 360 s and T = 40 °C: 1. potassium permanganate (1.0 × 10−3 mol L−1) + sodium hydroxide (0.24 mol L−1); 2. chlortetracycline (0.50 µg mL−1) + (1); 3. tetracycline (0.50 µg mL−1) + (1); 4. oxytetracycline (0.25 µg mL−1) + (1). | ||
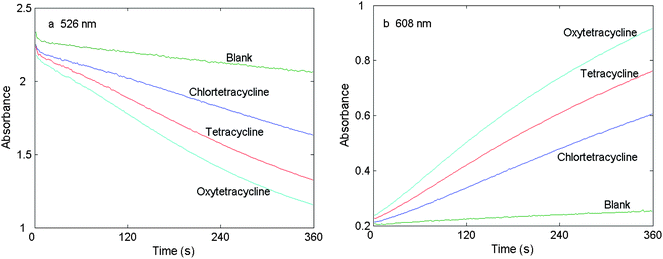 | ||
| Fig. 2 Absorbance versus time plot for chlortetracycline, tetracycline, oxytetracycline and the reagent blank. cNaOH = 0.24 mol L−1, cKMnO4 = 1.0 × 10−3 mol L−1, cCTC = cTC = cOTC = 0.25 µg mL−1, T = 40 °C and t = 360 s. | ||
Optimization of experimental conditions
In order to obtain an optimized reaction system, various experimental parameters have been investigated. The reagent concentrations and reaction conditions were optimized one at a time while setting other parameters constant. Each experiment was repeated three times and the concentrations of tetracyclines were kept at 0.25 µg mL−1.The effect of alkalinity of the reaction medium was investigated by performing the reaction with sodium hydroxide in the concentration range of 0.04–0.32 mol L−1. It was noticed that, when the sodium hydroxide concentration increased, the absorbance difference for tetracyclines increased substantially up to 0.24 mol L−1. In this work, 0.24 mol L−1 sodium hydroxide was selected as the optimum.
The effect of potassium permanganate was studied in the range of 0.2–1.6 × 10−3 mol L−1. The absorbance of tetracyclines increased substantially with the increase in potassium permanganate up to 1.0 × 10−3 mol L−1. Therefore, a potassium permanganate concentration of 1.0 × 10−3 mol L−1 was selected as the optimum concentration on basis of higher sensitivity.
According to Arrhenius' theory, the rate of a kinetic reaction mainly depends on the temperature.37 The effect of temperature on the reaction rate was studied in the range of 20–60 °C. The absorbance difference for the three tetracyclines increased with the increasing temperature up to 40 °C. Therefore, 40 °C was chosen as the optimum.
Linear calibration models for single component
A set of samples with different concentrations for chlortetracycline, tetracycline and oxytetracycline was prepared, and the kinetic measurements at 526 and 608 nm were carried out under the optimum conditions. Kinetic data for each compound with different concentrations are presented in Fig. 3, and the statistical results and the evaluated parameters of the linear equations for these three tetracyclines are listed in Table 1.38 The linear concentration range at both peak wavelengths is 0.05–0.75 µg mL−1 for chlortetracycline and tetracycline and 0.02–0.34 µg mL−1 for oxytetracycline, respectively. The limits of detection (LODs) were 0.011 µg mL−1 (526 nm) and 0.012 µg mL−1 (608 nm) for chlortetracycline; 0.013 µg mL−1 (526nm) and 0.028 µg mL−1 (608 nm) for tetracycline; and 0.015 µg mL−1 (526 nm) and 0.027 µg mL−1 (608 nm) for oxytetracycline. It is clear that parameters from the data collected at 526 nm are better when compared with those from 608 nm.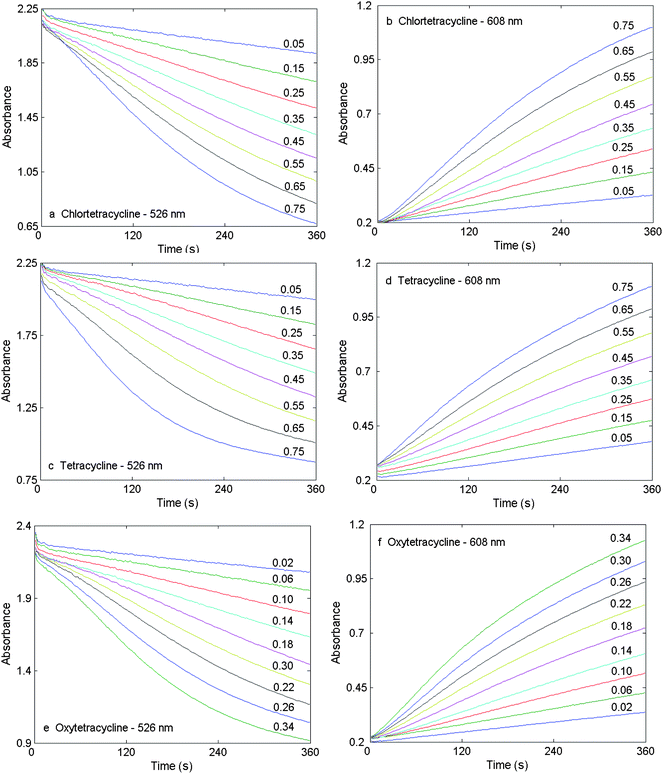 | ||
| Fig. 3 Kinetic curves for chlortetracycline, tetracycline and oxytetracycline with different concentrations (µg mL−1) at 526 and 608 nm. Experimental conditions are as in Fig. 2. | ||
| Parametersa | Chlortetracycline | Tetracycline | oxytetracycline | |||
|---|---|---|---|---|---|---|
| 526 nm | 608 nm | 526 nm | 608 nm | 526 nm | 608 nm | |
| a s I , sS, sR and LOD are the standard deviation of the intercept, the standard deviation of the slope, the standard deviation of the regression and the limit of detection, respectively (Miller38). | ||||||
| Number of samples | 8 | 8 | 8 | 8 | 9 | 9 |
| Linear range (mg L−1) | 0.05–0.75 | 0.05–0.75 | 0.05–0.75 | 0.05–0.75 | 0.02–0.34 | 0.02–0.34 |
| Correlation coefficient | 0.9999 | 0.9994 | 0.9998 | 0.9994 | 0.9995 | 0.9994 |
| Intercept | 2.035 | 0.261 | 2.107 | 0.318 | 2.205 | 0.273 |
| Slope (L mg−1) | 1.671 | 1.105 | 1.600 | 1.021 | 3.561 | 2.515 |
| s I | 0.005 | 0.007 | 0.006 | 0.007 | 0.009 | 0.007 |
| s S | 0.011 | 0.016 | 0.012 | 0.014 | 0.043 | 0.032 |
| s R | 0.007 | 0.010 | 0.008 | 0.009 | 0.013 | 0.010 |
| LOD (mg L−1) | 0.013 | 0.028 | 0.015 | 0.027 | 0.011 | 0.012 |
Recently, some sensitive fluorescence methods for simultaneous determination of tetracyclines were reported. With PLS and fluorescence technique, the linear ranges and the LODs were 0.08–4.0 µg mL−1, 0.032 µg mL−1 for oxytetracycline, 0.08–1.5 µg mL−1 and 0.005 µg mL−1 for chlortetracycline and 0.1–4.0 µg mL−1 and 0.014 µg mL−1 for tetracycline, respectively.18 For simultaneous luminescent determination of tetracyclines, the tetracycline and oxytetracycline were assayed with the linear range of 0.03–0.35 µg mL−1 and LOD of 0.0065 µg mL−1.17 In another study, PARAFAC was used to interpret the fluorescence spectra for the determination of tetracycline, and the LOD was 0.013 µg mL−1.16 Therefore, the proposed method in this work has similar or even better sensitivity, and is therefore comparable with the reported methods.
Prediction of tetracyclines in synthetic mixtures
The orthogonal array design method was used to optimize the calibration set. Application of a four-level orthogonal array design, denoted by OA16(45),39 showed that sixteen standard samples (Table 2) were required. These calibration samples were analyzed by the kinetic method according to the experimental procedure previously described, and a 16 (standard solutions) × 181 (time points from 0 to 360 s with an interval of 2 s) two-way kinetic matrix (at 526 or 608 nm) and a 16 (standard solutions) × 401 (wavelengths) × 37 (time points of 0–360 s every 10 s) three-way kinetic-spectral matrix were obtained. Then PCR, PLS and RBF-ANN were used to resolve the two-way matrix, and NPLS, UPLS and UPCR were applied to a three-way data matrix, and different calibration models were established. For the RBF-ANN calibration models optimization is often non-trivial and many parameters require estimation. Overfitting has to be controlled such that the model fits the signal but excludes noise, otherwise predictions of atypical objects beyond calibration limits may occur. A model may be tested with data during training; this will provide information on how much training is required in order for the model to perform well without overfitting. The testing can be carried out continuously by sequential comparison of the mean square error of the test data, and when this parameter stops improving, the training can cease. Thus, in this work, four ANN parameters—the mean squared error goal, spread coefficient, hidden layer and number of neurons, were optimized in this manner, and their values were 1 × 10−3, 20, 1 and 10, respectively. Then, to verify and evaluate the prediction ability of these established RBF-ANN and other calibration models, a verification set of another sixteen samples (Table 2) was prepared and analyzed by the proposed kinetic method. Table 3 shows the RPES, RPET and the percentage mean recovery obtained by applying the different chemometrics methods to the simultaneous determination of chlortetracycline, tetracycline and oxytetracycline in synthetic samples. In general, the %Recovery was better than about 85% for all the models tested with many results close to or better than 95%. Thus, from this perspective all methods have perfomed satisfactorily. As shown in Table 3, the results in terms of %RPES and %RPET obtained by the RBF-ANN model for the two-way data collected at 526 nm or 608 nm were better than those obtained by the PCR and PLS models. Moreover, for the two-way data, better predictions were obtained from the data collected at 526 nm than those at 608 nm, probably because the rate constants of the tetracyclines at 526 nm were larger (Section: Reaction kinetics of tetracyclines). Given that the %RPET for the RBF-ANN% (526 nm) was about 6 and %RPES values for all three analytes were below 10,40 it would seem that with the data collected at 526 nm, a properly calibrated RBF-ANN model is the method of choice for the prediction of the three analytes simultaneously. PCR and PLS models (λ = 526 nm) have % RPET below 10 but on the otherhand the %RPES for chlortetracycline, tetracycline were well above this number. In comparison with the discussion above, all three—way calibration models did not perform well with the %RPET 19 and the range of the %RPES between ∼15 and 24, and thus cannot be regarded as suitable for this analysis.| Sample number | Calibration set | Prediction set | ||||
|---|---|---|---|---|---|---|
| CTC | TC | OTC | CTC | TC | OTC | |
| 1 | 0.10 | 0.10 | 0.18 | 0.15 | 0.15 | 0.16 |
| 2 | 0.10 | 0.20 | 0.08 | 0.15 | 0.25 | 0.10 |
| 3 | 0.10 | 0.30 | 0.12 | 0.15 | 0.35 | 0.14 |
| 4 | 0.10 | 0.45 | 0.04 | 0.15 | 0.40 | 0.06 |
| 5 | 0.20 | 0.10 | 0.08 | 0.25 | 0.15 | 0.10 |
| 6 | 0.20 | 0.20 | 0.18 | 0.25 | 0.25 | 0.16 |
| 7 | 0.20 | 0.30 | 0.04 | 0.25 | 0.35 | 0.06 |
| 8 | 0.20 | 0.45 | 0.12 | 0.25 | 0.40 | 0.14 |
| 9 | 0.30 | 0.10 | 0.12 | 0.35 | 0.15 | 0.14 |
| 10 | 0.30 | 0.20 | 0.04 | 0.35 | 0.25 | 0.06 |
| 11 | 0.30 | 0.30 | 0.18 | 0.35 | 0.35 | 0.16 |
| 12 | 0.30 | 0.45 | 0.08 | 0.35 | 0.40 | 0.10 |
| 13 | 0.45 | 0.10 | 0.04 | 0.40 | 0.15 | 0.06 |
| 14 | 0.45 | 0.20 | 0.12 | 0.40 | 0.25 | 0.14 |
| 15 | 0.45 | 0.30 | 0.08 | 0.40 | 0.35 | 0.10 |
| 16 | 0.45 | 0.45 | 0.18 | 0.40 | 0.40 | 0.16 |
| Data sources and chemometrics method | %RPESa | %RPETa | ||
|---|---|---|---|---|
| Chlortetracycline | Tetracycline | Oxytetracycline | ||
| a Relative prediction error (single component): RPES = 100 × [∑ni=1(cij(found) − cij(added))2/∑ni=1(cij(added))2]0.5, and total relative prediction error: RPET = 100 × [∑ni=1∑mj=1(cij(found) − cij(added))2/∑ni=1∑mj=1(cij(added))2]0.5. b Values correspond to the number of factors used. c Parameter values are: mean squared error goal, nodes in the hidden layer and the spread coefficient, respectively. d Mean recoveries (%), %Recovery = 100 × ∑ni=1[cij(found)/cij(added)]/n. | ||||
| Two-way kinetic data from 526 nm | ||||
| PCR (5)b | 8.7 (94.8)d | 6.4 (97.6) | 16.6 (105) | 8.6 |
| PLS (5)b | 4.1 (99.4) | 7.4 (98.4) | 13.3 (97.4) | 6.8 |
| RBF-ANN (10−3, 10, 20)c | 4.7 (98.0) | 7.2 (102.2) | 3.9 (97.0) | 5.9 |
| Two-way kinetic data from 608 nm | ||||
| PCR (5)b | 8.2 (99.9)d | 12.4 (103) | 19.2 (87.8) | 11.4 |
| PLS (5)b | 6.8 (99.7) | 12.3 (103) | 17.4 (88.8) | 10.6 |
| RBF-ANN (10−3, 8, 60)c | 6.9 (98.7) | 9.1 (102) | 14.1 (91.8) | 8.7 |
| Three-way kinetic-spectra data from 325–725 nm within 0–360 s | ||||
| NPLS (4)b | 15.9 (86.3) | 23.3 (91.6) | 20.2 (90.6) | 19.9 |
| UPCR (6)b | 15.8 (86.3) | 22.3 (90.8) | 19.5 (89.6) | 19.3 |
| UPLS (6)b | 15.8 (86.2) | 21.2 (90.0) | 18.7 (88.7) | 18.6 |
Interference study
To investigate the selectivity of the proposed method, the effect of various common substances and ions which may exist in the samples, was tested on mixtures of the analytes (0.3 µg mL−1 of each TC). The tolerance limits were taken as the concentrations (µg mL−1), which resulted in a relative error of 10% or less in the concentration of analytes. The tolerance limits were: 400-fold for glucose; 200-fold for starch; 260-fold for citric acid; 100-fold for Na+, Mg2+, Ca2+, Fe2+, Ni2+ and Pb2+; 0.5-fold for Al3+ and Zn2+.Analysis of animal feed and muscle samples: An application
To evaluate the proposed kinetic method, the chlortetracycline, tetracycline and oxytetracycline in feed and muscle samples was treated as described (Section: Preparation of samples), and then 100 µL of the extract were transferred to the spectrophotometric cell for analysis. The RBF-ANN calibration based on the kinetic data collected at 526 nm, as described above, was applied for the resolution of the mixtures of chlortetracycline, tetracycline and oxytetracycline in feed and muscle samples. In addition, the HPLC method was used as a reference method to compare the performance of the proposed method. The results obtained from the proposed kinetic method showed good agreement with that from the HPLC measurements (Table 4). The difference between the %Recovery of the proposed methods and HPLC, for quantification of each analytes in muscle sample, was less than 5% for chlortetracycline, 1.5% for tetracycline and 2% for oxytetracycline. It is also evident that the %Recovery of each analyte in feed samples were in the range from 93%–105%. These results strongly suggest that the differential kinetic method with the aid of chemometrics presented in this paper, under established experimental conditions, could be successfully applied to the simultaneous determination of chlortetracycline, tetracycline and oxytetracycline in feed and muscle samples.| Samples | Added/mg g−1 | Measured/mg g−1 | Recovery (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| CTC | TC | OTC | CTC | TC | OTC | CTC | TC | OTC | |
| RBF-ANN for feed samples | |||||||||
| 1 | 0.025 | 0.025 | 0.025 | 0.024 | 0.023 | 0.026 | 96.9 | 93.6 | 104.9 |
| 2 | 0.050 | 0.050 | 0.050 | 0.049 | 0.047 | 0.050 | 98.2 | 94.4 | 100.8 |
| 3 | 0.100 | 0.100 | 0.100 | 0.100 | 0.099 | 0.099 | 100.2 | 99.3 | 98.6 |
| RBF-ANN for muscle samples | |||||||||
| 1 | 0.005 | 0.005 | 0.005 | 0.0044 | 0.0046 | 0.0047 | 89.1 | 91.3 | 93.0 |
| 2 | 0.010 | 0.010 | 0.010 | 0.0088 | 0.0093 | 0.0094 | 88.2 | 93.2 | 94.2 |
| 3 | 0.020 | 0.020 | 0.020 | 0.0181 | 0.0191 | 0.0192 | 90.4 | 95.3 | 96.0 |
| HPLC for muscle samples | |||||||||
| 1 | 0.005 | 0.005 | 0.005 | 0.0047 | 0.0045 | 0.0047 | 93.8 | 90.0 | 94.6 |
| 2 | 0.010 | 0.010 | 0.010 | 0.0089 | 0.0094 | 0.0094 | 89.2 | 94.0 | 94.0 |
| 3 | 0.020 | 0.020 | 0.020 | 0.0184 | 0.0193 | 0.0194 | 92.0 | 96.5 | 97.2 |
Conclusion
A sensitive spectrophotometric kinetic method for simultaneous determination of chlortetracycline, tetracycline and oxytetracycline in mixtures has been researched and developed. Chemometrics methods were required to obtain a suitable calibration model for the simultaneous prediction of the analytes. Of the several methods attempted, the RBF-ANN calibration performed best on the basis of %RPES (Range: 4–7) and %RPET (6). The proposed method was applied for the determination of the tetracyclines in spiked muscle and animal feed samples, and was verified with the use of the HPLC method. The results were satisfactory on the basis of %Recovery and the HPLC verification trial. This method therefore, is a potential alternative to the commonly used HPLC approach, and has the advantages of relatively high selectivity, low-cost, easily available chemicals and instrumentation.Acknowledgements
The authors gratefully acknowledge the financial support of this study by the Natural Science Foundation of China (No. NSFC20562009), and the State Key Laboratory of Food Science and Technology of Nanchang University (SKLF-MB-200807 and SKLF-TS-200919).References
- S. Bogialli, R. Curini, A. Di Corcia, A. Lagana and G. Rizzuti, J. Agric. Food Chem., 2006, 54, 1564–1570 CrossRef CAS.
- P. Vinas, N. Balsalobre, C. Lopez Erroz and M. Hernandez Cordoba, J. Chromatogr., A, 2004, 1022, 125–129 CrossRef CAS.
- J. M. Traviesa Alvarez, J. M. Costa Fernandez, R. Pereiro and A. Sanz Medel, Anal. Chim. Acta, 2007, 589, 51–58 CrossRef CAS.
- P. Masawat and J. M. Slater, Sens. Actuators, B, 2007, 124, 127–132 CrossRef.
- U.S. Code of Federal Regulations, Title 21, Part 150/556/720, Section 500, U.S. Government Printing Office, Washington DC, 2008, (Chapter 1). http://www.accessdata.fda.gov Search PubMed.
- The European Agency for the Evaluation of Medicinal Products, Committee for Veterinary Medicinal Products. Oxytetracycline, Tetracycline, Chlortetracycline, Summary Report (3), EMEA/MRL/023/95. http://www.emea.europa.eu/pdfs/vet/mrls/Tetracyclines(3)02395.pdf.
- S. W. Yang, J. M. Cha and K. Carlson, J. Chromatogr., A, 2005, 1097, 40–53 CrossRef CAS.
- J. W. Fritz and Y. Zuo, Food Chem., 2007, 105, 1297–1301 CrossRef CAS.
- A. Crecelius, M. R. Clench, D. S. Richards and V. Parr, J. Chromatogr., A, 2002, 958, 249–260 CrossRef CAS.
- J. J. Pesek, M. T. Matyska, T. Bloomquist and G. Carlon, J. Liq. Chromatogr. Relat. Technol., 2005, 28, 3015–3024 CrossRef CAS.
- A. Townshend, W. Ruengsitagoon, C. Thongpoon and S. Liawruangrath, Anal. Chim. Acta, 2005, 541, 103–111 CrossRef.
- L. Nozal, L. Arce, B. M. Simonet, A. Rios and M. Valcarcel, Anal. Chim. Acta, 2004, 517, 89–94 CrossRef CAS.
- A. L. Myllyniemi, L. Nuotio, E. Lindfors and H. Korkeala, Analyst, 2004, 129, 265–269 RSC.
- D. S. Aga, R. Goldfish and P. Kulshrestha, Analyst, 2003, 128, 658–662 RSC.
- P. Izquierdo, A. Gomez Hens and D. Perez Bendito, Anal. Chim. Acta, 1994, 292, 133–139 CrossRef CAS.
- N. Rodriguez, B. D. Real, M. C. Ortiz, L. A. Sarabia and A. Herrero, Anal. Chim. Acta, 2009, 632, 42–51 CrossRef CAS.
- G. A. Ibanez, Talanta, 2008, 75, 1028–1034 CrossRef CAS.
- R. S. Valverde, M. D. G. Garcia, M. M. Galera and H. C. Goicoechea, Anal. Chim. Acta, 2006, 562, 85–93 CrossRef.
- R. S. Valverde, M. D. G. Garcia, M. M. Galera and H. C. Goicoechea, Talanta, 2006, 70, 774–783 CrossRef CAS.
- B. M. Quencer and S. R. Crouch, Crit. Rev. Anal. Chem., 1993, 24, 243–262 CAS.
- M. Blanco, J. Coello, H. Itrurriaga, S. Maspoch and M. Redon, Appl. Spectrosc., 1994, 48, 37–43 CAS.
- G. Lopez Cueto, J. F. Rodriguez Medina and C. Ubide, Analyst, 1997, 122, 519–523 RSC.
- S. Ventura, M. Silva, D. Perez Bendito and C. Hervas, Anal. Chem., 1995, 67, 1521–1525 CrossRef CAS.
- T. F. Cullen and S. R. Crouch, Mikrochim. Acta, 1997, 126, 1–9 CAS.
- D. T. Harvey, Modern Analytical Chemistry. New York: McGraw-Hill, 1999 Search PubMed.
- I. T. Jolliffe, Principal Component Analysis. New York: Springer, 1986 Search PubMed.
- A. Lorber, L. E. Wangen and B. R. Kowalski, J. Chemom., 1987, 1, 19–31 CAS.
- P. Geladi and B. R. Kowalski, Anal. Chim. Acta, 1986, 185, 1–17 CrossRef CAS.
- D. M. Haaland and E. V. Thomas, Anal. Chem., 1988, 60, 1193–1202 CrossRef CAS.
- Y. H. Pao, Adaptive Pattern Recognition and Neural Networks, Addison-Wesley, Reading, MA, 1989 Search PubMed.
- R. Bro, J. Chemom., 1996, 10, 47–61 CrossRef CAS.
- S. Wold, P. Geladi, K. Esbensem and J. Ohman, J. Chemom., 1987, 1, 41–56 CAS.
- A. C. Olivieri, J. Chemom., 2005, 19, 253–265 CrossRef.
- J. W. Batista Braga, R. Lajarim Cameiro and R. J. Poppi, Chemom. Intell. Lab. Syst., 2010, 100, 99–109 CrossRef CAS.
- GB/T 5009.116-2003, Determination of oxytetracycline, tetracycline and chlortetracycline residues in meat (HPLC).
- N. R. Draper and H. Smith, Applied Regression Analysis (3rd ed.). New York: Wiley, 1998 Search PubMed.
- R. Kellner, J. M. Mermet, M. Otto and H. M. Widmer, Analytical Chemistry. New York: Wiley-VCH, 1998 Search PubMed.
- J. N. Miller, Analyst, 1991, 116, 3–14 RSC.
- Y. N. Ni, The application of Chemometrics in Analytical Chemistry; Beijing: Chinese Science Press, 2004; p 69–83 Search PubMed.
- Y. N. Ni and S. Kokot, Anal. Chim. Acta, 2008, 626, 130–146 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |