A microfluidic device for tissue biopsy culture and interrogation
Abigail
Webster
a,
Charlotte E.
Dyer
b,
Stephen J.
Haswell
a and
John
Greenman
*b
aDepartment of Chemistry, University of Hull, Cottingham Rd, Hull, UK
bDivision of Cancer, Postgraduate Medical Institute, University of Hull, Hull, UK. E-mail: j.greenman@hull.ac.uk; Fax: +44 (0)1482466996; Tel: +44 (0)1482466032
First published on 5th July 2010
Abstract
This communication reports the development of a microfluidic device capable of maintaining the long-term culture of viable tissue biopsies. Tissue-based models will enable evaluation of cell–cell and cell–matrix interactions within multi-cellular systems. The device demonstrated is a prototype, fabricated with the capacity to receive biopsy samples up to 2 mm3, from various tissue sources. Presently, this system has been tested with human colorectal tissue biopsies, for periods in excess of 3 days. The response of normal colorectal tissue and neoplastic biopsies to hypoxia was assayed by the release of vascular endothelial growth factor (VEGF) into the media, which was measured off-chip. As anticipated, the hypoxia induced a greater VEGF response in the tumour biopsies than the non-malignant tissue.
The application of microfluidic devices to the analysis of biological samples is a rapidly developing field. Culturing tissue biopsies in a microfluidic device will provide a more holistic model for detecting cellular response to changes, such as drug stimuli, because the device can accurately reproduce many of the key parameters of the in vivo environment. Compared with a traditional cell culture model, biopsy tissue retains the complex cell–cell and cell–matrix communication/interaction; furthermore tissue biopsies are far less likely to undergo surface marker changes, a phenomenon that occurs over time with cell culture as essentially a portion of the tissue in its native state is being studied. Advances in 3D culturing1 and tumour spheroid models2 have been reported recently as it is believed these will provide superior models of tumour response to anticancer drugs in preclinical in vitro experiments. Whilst a great deal of literature describes cell culture within microfluidic devices,3,4 relatively few report whole tissue based methodology due to the difficulty in culturing this type of sample. Previous work within this area has largely been restricted to culturing ultra-thin brain tissue slices in polydimethylsiloxane (PDMS) devices for a maximum of 3 h.5–7 Culturing tissue biopsies, not cells, in excess of 24 hours would generally be considered long-term.8
The most important factors when culturing cells or tissue are temperature, sterility, nutrients (via media) and gas supply. Gas saturation of media is essential as media require pH stabilisation using CO2, while gas concentrations, especially O2, are vital to the normal functioning of cellular processes. Tissue hypoxia, resulting from inadequate oxygen supply, compromises cellular functions and in tumours can be described as a pathophysiologic consequence of a functionally and structurally disturbed microcirculation. Hypoxia not only affects tumour cells directly, but is also an important parameter to be considered when investigating drug treatments, i.e. hypoxic regions within the tumour microenvironment will alter the local pH and drug sensitivity.9 During cancer progression, the neoplastic tissue grows too large for diffusion to supply the nutrient needs of the inner cell mass. The neoplastic tissue releases a variety of factors including vascular endothelial growth factor (VEGF), a fundamental neovascularisation factor,10 which is required to stimulate angiogenesis and thus create new supply routes that will provide the rapidly dividing tumour mass with essential nutrients. Overexpression of VEGF has been demonstrated in almost all solid tumours including colonic neoplasms.11 Critically low gaseous supply can be generated in the laboratory to mimic the process by creating a hypoxic environment, and tumours release VEGF in response to this stimulation.12 Hence, VEGF is a useful biomarker for monitoring the response of normal and neoplastic tissue biopsies to changes in the microenvironment within the tissue cavity of the prototype device due to changes in the gassing regime.
Results and discussion
The microfluidic device was designed using AutoCAD software and was fabricated in-house according to published procedures13 with some adaptation. Briefly, the channel network (50 µm deep) was fabricated by photolithography on B270 super white crown glass (1 mm thick). A 3 mm top-plate was predrilled with 1.25 mm diameter holes to accommodate the tubing and a central 3 mm tissue cavity (Fig. 1A). The two plates were then thermally bonded in a muffle furnace. A flat bottomed nanoport assembly with an internal diameter of 6.4 mm (Upchurch Scientific, UK) was adhered to the top-plate over the tissue cavity, using thermally activated glue according to the manufacturer's instructions, to enable easy access for insertion or removal of tissue biopsies (Fig. 1B).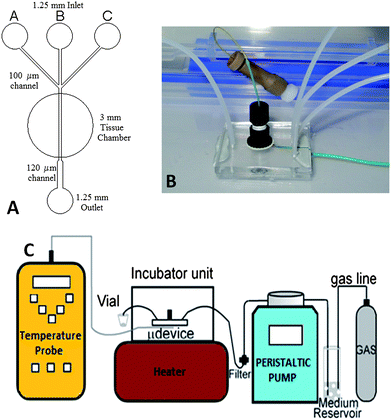 | ||
| Fig. 1 (A) Channel schematic of the device. (B) Photograph showing the glass microdevice with attached nanoport. (C) General schematic of the assembly showing the pumping system, gassing to media reservoir and flow of the system. | ||
The experimental assembly offers a high degree of fluidic and gas saturation control and the potential for manipulating the tissue microenvironment. The device was attached to a peristaltic pump (Minipuls 3, Gilson, France) that provided in-line filtered (Millipore 0.22 µm PES membrane, Millipore Ireland), gas-saturated media of known concentrations (Fig. 1C). A 30 ml reservoir of Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% (v/v) fetal calf serum and penicillin/streptomycin (10 unit per ml) (Invitrogen, UK) was gassed with 75% O2/20% N2/5% CO2. Prior to each experiment, a microdevice was primed with this medium.
Temperature was kept at a constant 37.5 ± 0.2 °C using a hotplate (Stuart CB160), monitored with an optical temperature probe (OpSens, Hart Scientific, UK) located directly under the tissue chamber. pH stabilisation of the media was achieved using 5% CO2 in both gas mixtures and was verified by measuring the pH of the media after it had flowed through an empty, sterile microdevice. The pH was stable at 7.4 when no tissue was present in the device, minor fluctuations in pH were seen when the device contained tissue, which was most probably due to changes caused by the tissue and the factors released.
Tissue was taken following ethics approval from Hull and East Yorkshire Local Research Ethics Committee 07/H1304/105 and NHS Trust R0568. Colorectal tissue biopsies, normal and neoplastic, were sectioned and a sample weighing approx. 5–10 mg was then placed in the device. The control was tissue taken from the distal region of the tumour biopsy. The tissue was perfused with supplemented DMEM at a flow rate of 1 µl min−1. After a period of normoxia (18 h), this was replaced by perfusion with an aliquot of supplemented DMEM saturated with 95% N2/5% CO2, at the same flow rate, to generate a hypoxic environment. After approximately 26 h the medium was changed to supplemented DMEM that had been O2 purged and normoxia was restored for 22 h, finishing with a second (4 h) period of hypoxia. During this time, aliquots of the supernatant were collected at 2 h intervals after flow over the tissue, however overnight samples were collected for a 16 h period. The supernatant was frozen immediately after collection and analysed for VEGF release using a commercial ELISA (R&D Systems, UK). Samples were measured in triplicate and the results were normalised for VEGF concentration per mg of tissue.
The tumour tissue responded to the hypoxic conditions of the gas regime with an enhanced production of VEGF, as measured by ELISA. The normal tissue also responded with a release of VEGF, but at lower levels, which is in accord with the expected in vivo response as colorectal neoplastic tissue is known to overexpress VEGF.11 The release of VEGF was reduced when the gaseous microenvironment was restored to normoxia, and subsequently a reduction in the amount of VEGF was seen in the supernatant, these results are shown in Fig. 2.
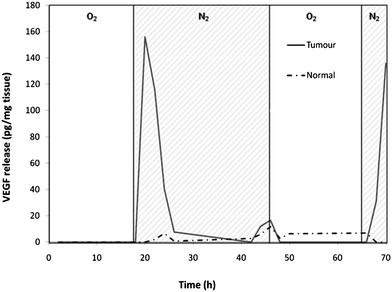 | ||
| Fig. 2 Results from the ELISA showing the expression of VEGF in response to changes in the gas regime. The tumour tissue shows a greater increase of VEGF release than seen with the normal tissue after the hypoxic period (N2/CO2 gassed media). Hypoxic periods are indicated by cross-hatched N2 areas on the graph. Data shown are representative of 3 similar experiments. | ||
Summary
Initial testing of this experimental system has shown adequate gas saturation and sterile media delivery, which enables the long-term culturing of tissue. It is expected that the microfluidic device will now be used to investigate chemotherapeutic drugs that prevent proliferation and/or induce apoptosis by assaying VEGF and other relevant biomarkers. For the majority of in vitro studies, apart from those on blood cells, tissues are disaggregated and cultured as individual cell populations, with the concomitant loss of cell organisation and interaction with a tissue-specific extracellular matrix. With the advent of tissue engineering the complexity and importance of the 3D structure has become very apparent for normal14 and disease processes.15 Furthermore, it is acknowledged that many of the 3D models or co-cultures cannot yet represent the natural or pathophysiological state, although many studies are in progress to address this problem.16 The simplistic and flexible design of the current system offers a device that will be capable of real time analytical interrogation of various tissue types, supporting a variety of diverse applications in which a microfluidic tissue device can be used to guide drug treatment selection in a clinical setting. This will be particularly valuable for diseases where a cocktail of drugs are required, for example in cancer therapy. This research is timely in driving towards less animal-reliant testing in the pharmaceutical industry and more personalised care in the medical arena. It also offers the potential for development of point-of-care units capable of assessing the suitability of drug treatments prior to administration to patients, providing more cost effective and, more importantly, clinically effective treatments.Acknowledgements
We gratefully acknowledge the support of the BBSRC (Grant no. BB/E002722/1), members of the Academic Surgical Unit for tissue biopsies and Dr Leigh Madden for invaluable discussions and advice.References
- J. Vukasinovic, D. K. Cullen, M. C. Laplaca and A. Glezer, Biomed. Microdevices, 2009, 11, 1155–1165 CrossRef.
- Y. S. Torisawa, A. Takagi, Y. Nashimoto, T. Yasukawa, H. Shiku and T. Matsue, Biomaterials, 2007, 28, 559–566 CrossRef CAS.
- B. G. Chung, J. W. Park, J. S. Hu, C. Huang, E. S. Monuki and N. L. Jeon, BMC Biotechnol., 2007, 7, 60 CrossRef.
- J. H. Wittig, A. F. Ryan and P. M. Asbeck, J. Neurosci. Methods, 2005, 144, 79–89 CrossRef.
- A. J. Blake, T. M. Pearce, N. S. Rao, S. M. Johnson and J. C. Williams, Lab Chip, 2007, 7, 842–849 RSC.
- Y. Choi, M. A. McClain, M. C. LaPlaca, A. B. Frazier and M. G. Allen, Biomed. Microdevices, 2007, 9, 7–13 CrossRef.
- P. A. Passeraub, A. C. Almeida and N. V. Thakor, Biomed. Microdevices, 2003, 5, 147–155 CrossRef CAS.
- S. M. Hattersley, C. E. Dyer, J. Greenman and S. J. Haswell, Lab Chip, 2008, 8, 1842–1846 RSC.
- O. Tredan, C. M. Galmarini, K. Patel and I. F. Tannock, J. Natl. Cancer Inst., 2007, 99, 1441–1454 CrossRef CAS.
- T. Tokunaga, Y. Oshika, Y. Abe, Y. Ozeki, S. Sadahiro, H. Kijima, T. Tsuchida, H. Yamazaki, Y. Ueyama, N. Tamaoki and M. Nakamura, Br. J. Cancer, 1998, 77, 998–1002 CAS.
- L. F. Brown, B. Berse, R. W. Jackman, K. Tognazzi, E. J. Manseau, H. F. Dvorak and D. R. Senger, Am. J. Pathol., 1993, 143, 1255–1262 Search PubMed.
- K. Toshio, K. Yasuhiko, T. Shinji, O. Seiji, M. Norimasa, K. Eijiro, I. Masanori and C. Kazuaki, Int. J. Cancer, 2003, 105, 176–181 CrossRef.
- T. McCreedy, Anal. Chim. Acta, 2001, 427, 39–43 CrossRef CAS.
- L. G. Griffith and A. S. Melody, Nat. Rev. Mol. Cell Biol., 2006, 7, 211–224 CrossRef CAS.
- P. Friedl and K. Wolf, Nat. Rev. Cancer, 2003, 3, 362–374 CrossRef CAS.
- M. P. Schwartz, B. D. Fairbanks, R. E. Rogers, R. Rangarajan, M. H. Zaman and K. S. Anseth, Integr. Biol., 2010, 2, 32–40 RSC.
| This journal is © The Royal Society of Chemistry 2010 |