Detection of apoptosis: A review of conventional and novel techniques
Michelle M.
Martinez
,
Randall D.
Reif
and
Dimitri
Pappas
*
Department of Chemistry and Biochemistry, Texas Tech University, Lubbock, TX 79409-1061, USA. E-mail: d.pappas@ttu.edu
First published on 12th July 2010
Abstract
Apoptosis, or programmed cell death, has been shown to play a role in a number of diseases, including heart disease and cancer. Apoptosis is also a vital cellular process that helps to regulate tissue growth, fetal development, immune response, and a host of other biological processes. Deviation from the careful regulation of apoptosis is responsible for a host of diseases and health concerns. As such, understanding the process of apoptosis is vital for therapy development. Over the years, a number of methods have been discovered and developed to detect apoptosis. There are several standard techniques such as electron microscopy, the TUNEL assay, and flow cytometry. In addition, new techniques are quickly emerging, such as microfluidic devices, single molecule spectroscopy, and electrochemical methods. This review will cover some examples of the most common techniques as well as some new techniques in order to show the broad spectrum of methods available to detect apoptosis in cells.
1. Introduction
Apoptosis—programmed cell death—is a carefully regulated process that is responsible for many biological processes such as normal cell turnover, proper development and functioning of the immune system, hormone-dependent atrophy, and embryonic and brain development.1 Like mitosis, it is a highly regulated cell process that arises from strictly conserved signals in healthy individuals. Apoptosis is the mechanism balancing cell turnover in proliferating/regenerating tissue such as intestinal lining and skin. Apoptosis is also an organism's main method of selective cell removal; governing cell removal in processes as disparate as brain development and immune response. Apoptosis-like mechanisms are found in a wide variety of organisms, with many of the key regulatory proteins having homologs across species. Like other highly regulated cellular processes, apoptosis is characterized by specific morphological features and biochemical cascades. Many of the proteins that are critical to apoptosis have been identified; however, the molecular mechanisms of apoptotic induction and inhibition are not fully understood and are the focus of continued research.Since the apoptotic process is highly regulated, changes in those regulatory pathways lead to various forms of disease. In some cases, cell stress may induce unwanted apoptosis, such as in the case of many forms of heart disease. Apoptosis may occur during several stages of heart disease, resulting in deterioration of cardiac muscle over time. In addition, inhibition of caspases has been shown to reduce apoptosis in ischemia-perfusion in rat models,2 and can be used during cardiac infarcts to prevent extensive damage to myocardium. A breakdown in the apoptotic machinery can also lead to cancer. In this case, triggering apoptosis selectively is the ultimate goal of many researchers. Resistance to the apoptotic pathway—primarily via mutations in the p53 protein—make many anticancer drug candidates ineffective.3 Since apoptosis plays such a large role in both healthy organism function as well as diseases, understanding the mechanisms at the molecular level would provide a deeper insight into diseases and could influence advancements in treatment and diagnosis.4
2. The role of apoptosis in development and disease
There is a strong correlation with changes in the regulation of apoptosis and diseases in the body. When the equilibrium between cell growth and cell death is no longer regulated properly, there is dysfunction in tissue and subsequently the respective organ. Apoptosis is not only a mechanism to control cell number and tissue size, but also to remove infected, damaged, or stressed cells from the organism.5 However, there have been some viruses and bacteria that have developed the ability to inhibit apoptosis mechanisms. Certain cancers, such as high risk cervical cancer, is triggered by viral inhibition of apoptosis.6 Currently, an important target for cancer research is based on the understanding of the apoptosis machinery and resistance to apoptosis initiation. The targeted induction of apoptosis or blocking inhibitory mechanisms have been identified as therapeutic targets.7 Several types of cancer have been identified as the result of defects in the apoptosis machinery, such as B-cell chronic lymphocytic leukemia, follicular lymphoma, and tumors affected by T-cell leukemia/lymphoma virus-1.7Most cancers are associated with resistance to apoptosis initiation, specifically in p53 mutations. In other diseases, an increased rate of apoptosis has been implicated as a mechanism for tissue atrophy. For example, elevated rates of apoptosis have been identified as one mechanism for the loss of nerve cells in both strokes and Alzheimer's disease.8 Apoptosis has been shown to occur before necrosis in mouse models of muscular dystrophy,9 with a peak in apoptotic cells detected in 4-week-old mice. Interestingly, apoptosis markers (condensed nuclei, chromatin fragmentation) in that study were detected before morphological changes in muscle. As mentioned previously, a major source of cardiomyocyte atrophy is apoptosis. Since both the first10 and second leading causes of death in the developed world intricately involve apoptosis, it is vital to understand the process and also exert external regulation over it. An imbalance in either direction (increased or decreased apoptosis) can lead to different diseases, there is no single “catch all” therapeutic target. Therefore, understanding the mechanisms of apoptosis is crucial to combating many diseases.
3. Mechanisms of apoptosis
There are two main apoptotic pathways for nucleated cells; the extrinsic pathway (caspase 8 or death receptor pathway, Fig. 1A) and the intrinsic or mitochondrial pathway (caspase 9 pathway, Fig. 1B). Both pathways differ in how they are initiated; however, both converge to activation of the zymogen procaspase 3 into its active form, which leads to activation of additional, downstream caspases. Once activated, caspases cleave neighboring procaspases and initiate the caspase cascade. Ten major caspases are categorized into three major groups: initiators (caspase 2, 8, 9, 10), executioners (caspase 3, 6, 7), and inflammatory caspases (caspase 1, 4, 5). The initiator and executioner caspases are important in regulatory mechanisms and are hallmark indicators of apoptosis. Apoptosis events include DNA fragmentation, degradation of cytoskeletal and nuclear proteins, cross-linking of proteins, formation of apoptotic bodies, expression of ligands for phagocytic cell receptors, and finally uptake by phagocytic cells.11 The same apoptosis events are present in anucleated cells, except for nuclear condensation.12 For example, erythrocytes undergo eryptosis, a special form of apoptosis-like death that is primarily characterized by phoshatidylserine exposure and cell shrinkage. When these cells undergo oxidative stress, osmotic shock, or energy depletion, cyclooxygenase is activated and leads to the synthesis of prostaglandin E2, which in turn activates Ca2+. Calcium ion increase disrupts the balance of the Ca2+ and K+ channel which causes hyperpolarization and cell shrinkage. In platelets apoptotic-like events are activated by thrombin. Thrombin activates pro-apoptotic proteins that are translocated to the mitochondria. Once in the mitochondria, cytochrome c is released and caspase-9 is activated with the subsequent release of caspase-3. The process ends with phosphatidylserine exposure and cell shrinkage.12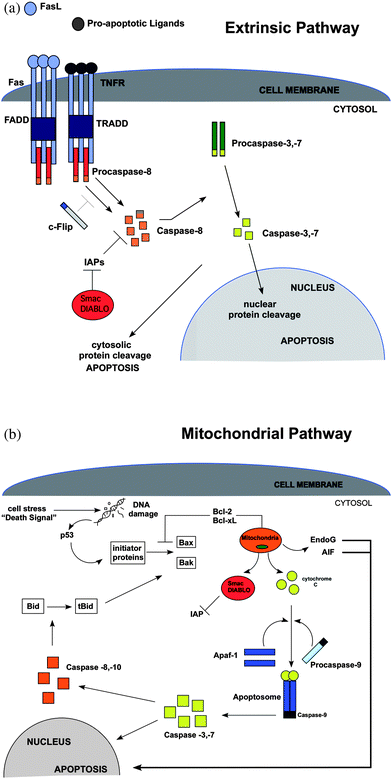 | ||
| Fig. 1 (A) The extrinsic or death receptor (DR) pathway. The intracellular portion of the DR is known as the death domain (DD). The complex that forms from the grouping of receptor–ligand complexes forms a death-inducing signaling complex (DISC), which recruits and assembles initiator caspase-8 and releases active caspase enzyme molecules into the cytosol. These molecules activate the effector caspases-3 and -7, resulting in nuclear protein cleavage and the initiation of apoptosis. FasL = Fas ligand; TNFR = tumor necrosis factor receptor; FADD = Fas-associated death domain; TRADD = TNF-associated death domain; C-FLIP = FLICE-like inhibitory protein. (B) The mitochondrial or intrinsic pathway. Activation in this pathway can occur through the initiator proteins upon induction of p53 by DNA damage, or through activation of Bax and Bak upon the conversion of Bid to tBid by caspase -8, -10. Activation at the mitochondrial membrane causes the release of several mitochondrial factors, such as cytochrome c which combines with Apaf-1 and procaspase-9 forming an apoptosome. Procaspase-9 is then converted into its active form, caspase-9, and then is able to activate caspase-3 or -7 allowing apoptosis to proceed. In addition, EndoG and AIF that stimulate apoptosis independent of caspases are also released. IAP = inhibitor of apoptosis; Apaf-1 = apoptosis activating factor-1; Bcl-2 and Bcl-xL = block the activation of Bax and Bak; Bcl-2 = B-cell lymphoma-2; Smac = second mitochondrial-derived activator of caspases; DIABLO = director inhibitor of apoptosis-binding protein with LOwpI; tBid = truncated Bid; EndoG = endonuclease G; AIF = apoptosis-inducing factor; Bax = Bcl-2-associated protein x; Bak = Bcl-2-associated protein k. | ||
Since apoptosis is present in certain tissues of developing and adult organisms, changes in apoptosis signaling must be discerned over a varying background. Elucidating the apoptosis process with high temporal resolution will enable new therapeutic targets aimed at either initiating or inhibiting apoptosis. Current methods are either incapable of making measurements with high temporal resolution, or are too invasive to use in dose–response studies. This review will provide an overview of the most common methods to detect apoptosis, as well as new techniques that have recently emerged. These emerging, enabling approaches hold a great deal of promise toward understanding apoptosis in greater detail and in new ways.
4. Measurement and detection approaches
4.1. Apoptosis detection by electron microscopy
The main advantage of electron microscopy lies in the high spatial resolution obtained during measurement. With environmental scanning electron microscopy, cell morphology is better preserved. Scanning electron microscopy is used to characterize morphological changes such as blebbing, and condensation of chromatin. If immunochemical staining is employed, then chemical information can be obtained in addition to spatial/morphological information. However, there are major limitations to using electron microscopy techniques.13 First, cells cannot be left viable after analysis, making any form of electron microscopy an “end point” detection. Second, some early stages of morphological changes can be detected; however, apoptotic cells that are detected by transmission electron microscopy (TEM) are in the last stage of apoptosis and therefore give no information to early stages of apoptosis. Third, electron microscopy can give rise to a high number of false positives. Finally, a great deal of time and a high skill level are needed in order to prepare and measure cells using electron microscopy.1 In 2003, Yasuhara et al. used electron microscopy to differentiate apoptosis from necrosis.14 Jurkat cells were incubated with staurosporin or a Fas ligand to induce apoptosis, or were exposed to extensive heat and N-ethylmaleimide (NEM) to induce necrosis. Control cells were treated with non-immune IgM. Eight hours after induction, the apoptotic group of cells was analyzed using TEM and showed signs of apoptosis morphology. These signs included intact organelles such as the mitochondria with signs of homogenous condensation of chromatin to one side of the nuclei. The inner matrix of some of the cells showed increased electron density. After 13 h and up to 24 h, the apoptotic cells showed signs of blebbing, apoptotic body formation, and fragmented nuclei; all hallmark signs of late stage apoptosis. After 8 h, the necrotic cells showed ruptured plasma membranes, irregular chromatin destruction, poorly stained cytoplasm, vacuole formation, and disrupted organelles.Electron microscopy has also been used to study rat cardiomyocytes, as well as other cell types. In the work by Roberg, the ultrastructural locations of cathespin D and cytochrome c were determined in cardiomyocytes that were exposed to oxidative stress.15 In their investigation, the location of both cytochrome c and cathespin D was determined at different times during apoptosis using several methods. Immunoelectron microscopy was able to detect the presence of cathepsin D in the cytosol 30 min after apoptosis induction and cytochrome c outside of the mitochondria 1 h after induction with naphthazarin. Temporal resolution was limited to the sampling time of 30 min per image, but could be increased.
4.2 Proteomic and genomic methods
While electron microscopy investigations yield morphological information, they provide only limited chemical information. Genomic and proteomic assays, measured using several different analytical methods, offer increased information content at the expense of the spatial resolution of electron microscopy. Like electron microscopy, most proteomic and genomic methods are invasive in that they cannot preserve cell viability.Methods focusing on DNA content or chromatin composition rely on the apoptotic mechanisms to degrade DNA in a uniform fashion. A common method capable of assaying the state of the cell chromatin is the DNA ladder assay. Breaks in chromatin result in the formation of DNA fragments, which can be detected by several methods. When detected by gel electrophoresis, the ladder formation of the fragmented DNA can be used to characterize apoptosis. In contrast, cells that are not apoptotic show no signs of ladder formation due to the lack of fragmentation. However, the DNA ladder assay offers low sensitivity.16 Because the ladder assay relies on the extent of oligonucleosomal cleavage, apoptosis cannot be detected until the later stages. Therefore, the ladder assay gives no information about the early stages of apoptosis, and the temporal resolution is low. In the work by Yasuhara et al., the authors used the comet assay, along with electron microscopy, to characterize apoptosis.14 Their results showed a distinct difference in the size of the comet head (i.e. intact chromatin), and the length of the tail between apoptotic cells and control cells. Unlike the ladder assay, the comet assay can detect various forms of DNA breakage. There are three main advantages to using the comet assay over the ladder assay and electron microscopy. These advantages are: higher sensitivity, more specific information about the extent of damage and heterogeneity of DNA, and ease of use.
Protein-based analyses assay the release of cytochrome c, up- or down-regulation of key inhibitory proteins, and the activation of caspases. Western blotting has been used extensively to detect cytochrome c as it is released from the mitochondria after induction of apoptosis via the intrinsic pathway. In the work by Roberg, cytochrome c release was detected after 30 min of treatment with the induction agent Naphthazarin.15 In this case, cytochrome c was detected in half the time required to detect its presence by immunoelectron microscopy. This is one of the earliest detections of apoptosis relative to the induction event. The benefit of measuring apoptosis reliably as early as possible (i.e. close to the induction event) is that both induction and inhibitory compounds can be assayed with greater temporal resolution. In addition, western blotting is sensitive and additional analyses can be performed on the detected proteins. Detection of activated proteins, or the absence of natural regulatory proteins in cancer, can also be performed by Western blotting and other gel-based methods. Like many separation methods, however, the progression of apoptosis cannot be followed for a given cell or sample of cells, since the technique is invasive. Therefore proteomic and genomic methods offer high information content, but cannot be used in intact cells, where apoptosis could potentially be both initiated and subsequently inhibited. However, the ability to detect the earliest stages of apoptosis will ensure that Western blotting will continue to play a key role in apoptosis-related research.
4.3 Spectroscopic techniques
Most spectroscopic techniques for apoptosis detection and measurement rely on fluorescence detection. Fluorescent probes and labels for apoptosis markers can be read out in a variety of formats such as flow cytometry, microscopy, microarray, etc. The application of flow cytometry is discussed in a later section, but the spectroscopic principles are covered here. Spectroscopic techniques can be used to detect immunochemical labels, or fluorescent probes that react to the cell environment during apoptosis. There are several spectroscopic techniques available to study apoptosis, including annexin V staining, the TUNEL assay, caspase detection, and measurement of mitochondrial membrane potential.Labeled annexin V can be applied in both flow cytometry and light microscopy to identify mid- to late-stage apoptotic cells. Koopman et al. were the first to introduce the report on the ability of annexin V to bind to the exposed phosphatidylserine on the surface of the cell membrane during apoptosis.17 In work done by van England and co-workers, an annexin V labeled assay was used to detect apoptosis in rat thymocytes treated with dexamethasone.18 After 4.5 h of exposure, the number of apoptotic cells increased from 4.1% in the control sample to 43.6%. The number of dead cells remained constant and was determined by propidium iodide (PI) staining.
Annexin-V was compared with a viability dye, calcein-AM, in work by Gatti et al.19 In this study the performance of annexin V-FITC and calcein-AM were compared for the detection of apoptosis in two different cell lines (PC 12 and NIH3T3) using confocal microscopy. Although annexin V-FITC was observed to be successful in detecting apoptosis, in one of the cell lines used by the authors (NIH3T3 cells), the method introduced bias based on the cell line and also the elapsed time after induction. The data showed that some of the NIH3T3 cells were late stage apoptotic based on calcein but were not annexin V-FITC positive. Therefore, annexin V positive cells may only represent a small portion of the cells that have entered the apoptotic pathway. When coupled with microscopy, calcein-AM can provide information on chromatin condensation, and give information about the function of the plasma membrane.20 If phosphatidylserine (PS) was lost at the early stages, it was not restored and could not be detected by annexin V labels. Therefore, in the NIH3T3 cell line, annexin V-FITC cannot be used as a reliable early marker for apoptosis. An additional caveat when using annexin-V is that the calcium content of the staining buffer is typically higher than physiological levels, and care must be taken to avoid artifacts introduced from the buffer alone.
Another common assay that detects apoptosis based on DNA fragmentation is by terminal deoxynucleotidyl transferase (Tdt)-mediated dUTP nick-end labeling (TUNEL assay). Using confocal microscopy, the detection of free 3′-OH DNA ends of nuclear condensed chromatin generated by apoptotic cells can be performed.1 Gavrieli and co-workers described developing the TUNEL assay for the study of apoptosis on a variety of tissues.21 Their results showed the arrangement of the DNA breaks observed by TUNEL were consistent with the expected arrangement of apoptosis. In addition, the TUNEL method provided in situ visualization of the DNA fragmentation process on a single-cell level. Their results showed the TUNEL method precedes the appearance of the ladder of nuclear fragments seen in gel electrophoresis and has the potential to give rise to ultrastructural aspects of the apoptosis process.
The TUNEL method can also be coupled with immunofluorescent labeling to increase information content. Ray et al. combined the TUNEL method with a double immunofluorescent labeling technique to study the fate of antigen presenting cells in experimental allergic encephalomyelitis (EAE) spinal cord tissue in rats.22 The authors modified the TUNEL assay by including an alkali-stable DIG-11-dUTP to subsequently detect the DIG-labeled DNA. The authors were able to not only confirm apoptosis in the spinal cord of rats with EAE, but were also able to determine the phenotype of the apoptotic cells responsible for the internucleosomally fragmented DNA.
More recently, Imasawa et al. investigated the role of a sphingolipid mediator, Sphingosine 1-phosphate (S1P).23 S1P is a bioactive lipid mediator that is critical in many biological processes, and acts through five cognate high-affinity receptors, (S1P1–S1P5). In previous studies, the authors found that S1P2 signaling leads to increased glucose uptake, and plays an important role in diabetes. According to the authors, studies have also shown that the presence of S1P at low concentrations can reduce apoptosis of pancreatic β-cells. Therefore, in their work, they examined the role of a S1P subtype, S1P2, in the progression of diabetes with pancreatic β-cells that had been induced with streptozotocin (STZ). In their results, the lack of S1P2 protected the mice islet cells from STZ-induced apoptosis. More specifically, islet cells that were TUNEL-positive were not only less abundant, but were 27% less abundant in S1P2 −/− males than in wild type (WT) male mice. Their findings concluded that the selective blockade of the sphingosine 1-phosphate subtype decreased the incidence of diabetes in mice.
Spectroscopic probes for membrane potential have been used for many years as early indicators of apoptosis. The opening of the mitochondrial permeability transition pore, followed by the release of cytochrome c into the cytosol, is one of the earliest steps in the intrinsic apoptotic pathway. Membrane potential has been shown as an indicator of this increased permeability, and can be tracked using several different dyes. There are many probes for membrane potential, although three that have been used extensively in apoptosis are JC-1,24 rhodamine 123,25 and mitotracker red.26 JC-1 forms red-fluorescent aggregates in intact mitochondria, but the aggregates disperse upon the loss of membrane potential. Tracking aggregates is therefore a simple approach for monitoring loss of membrane potential and subsequent apoptosis. Rhodamine 123 and mitotracker red sequester into the mitochondria, but this effect diminishes upon the loss of membrane potential. It is important when using these and other mitochondrial stains that the dye itself does not hinder metabolism of mitochondrial function. In addition, a secondary probe for apoptosis will be necessary to determine if the process concludes with cell death.
4.4 Flow cytometry
Flow cytometry is a common method used in conjunction with many other techniques in apoptosis detection or verification. While technically a spectroscopic technique, flow cytometry deserves mention in its own right. The high cell counts and multi-parameter detection of flow cytometry, combined with the possibility of sorting cells in some cases, makes the technique well suited to cell analysis. However, for apoptosis, flow cytometry can only measure a single point in time for a particular group of cells. In applications of apoptosis, flow cytometry gives information of the ratio of apoptotic cells and also measures cell viability in most cases. In addition, flow cytometry can be used to assay caspase activity, DNA cleavage, viability dyes, membrane dynamics, and a host of other apoptosis parameters such as light scatter. Dobrucki and Darzynkiweicz observed a decrease in forward and side scatter intensity due to cell shrinkage in early apoptotic cells.27 Since many labels and probes mentioned herein can be used with flow cytometry or other analytical methods, the major advantage of flow cytometry is cell throughput. Most flow cytometers can measure 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells in a few seconds whereas other methods, such as microscopy, can only measure a few dozen cells at a time in most cases.
000 cells in a few seconds whereas other methods, such as microscopy, can only measure a few dozen cells at a time in most cases.
In work by Yasuhara et al., apoptosis was differentiated from necrosis in Jurkat cells using annexin V-FITC staining.14 In addition, the cells were stained with propidium iodide (PI) for the detection of dead cells. Eight hours after induction with staurosporin and Fas ligand the percentage of cells that showed an apoptotic pattern were 2.36% for control, 19.87% for Fas ligand, 53.82% for staurosporin, 1.28% for NEM, and 0.08% for heat treatment, which confirmed apoptosis induction. In contrast, the percentage of dead cells using PI staining with annexin V-FITC in cells that were treated with NEM and heat were 97.46% for NEM and 99.01% for heat, which confirmed necrotic induced death.
Recently, Babu and co-workers used flow cytometry to test for apoptosis of a human monocyte cell line (THP-1). The authors utilized a group of cytotoxins (Stx-1) to induce apoptosis in the THP-1 cell line, and with an endpoint technique, the authors tested the stability of these cytotoxins.28 These toxins are the underlying cause for several serious conditions such as hemorrhagic colitis and hemolytic uremic syndrome (HUS). Their technique was based on the affinity of R-phycoerythrin-labeled annexin-V. The authors were interested in the stability of the cytotoxin in a food matrix at various temperatures and pH values. Their results showed that at higher temperatures and more acidic pH values, the toxin became inactive.
The design of new fluorescent dyes to monitor apoptosis using flow cytometry is a growing field of study. In a recent work by Wlodkowic and co-workers, new fluorescent probes were applied to the long term monitoring of apoptosis and the cell cycle.29 A number of cyanine SYTO probes were tested to assay caspase-dependent cell death. They discovered that one probe in particular, SYTO16, showed many properties of a dye useful for dynamic and high-throughput analysis. The dye was retained in the cells for long periods of time (72 h); the dye had little effect on the cell cycle, viability or migration, and there were few interferences with the assay readout. Cells stained with the dye were analyzed by both flow cytometry and microchip based cytometry. Such dyes have the potential to be powerful tools in automated drug screening and the analysis of samples with low cell numbers.
Virtually all fluorescence-based assays that do not rely on morphological detection can be analyzed with high cell counts by flow cytometry. For analyses where the same cells will be tracked over time, various forms of imaging cytometry and fluorescence microscopy are more suitable. Likewise, analyses where morphology is an indicator of apoptosis will also require some form of imaging detection. However, assays using annexin-V, DNA content, caspase activity, membrane potential, and other probes can be used with flow cytometry or flow sorting to achieve large cell counts and detect rare cells in a population.
4.5 Caspase activity assays
Caspases are a group of proteases that, when activated by apoptosis, act together as a cascade in the cleavage of proteins after aspartic acid residues.30 The ability of caspases to selectively cleave rather than randomly destroy proteins makes the cascade a good biochemical marker for detecting apoptosis. Hug and co-workers used a sensitive caspase assay to detect intracellular caspase activation by flow cytometry after induction of apoptosis in hematopoietic cells.31 In their assay, they introduced a new substrate, (Asp)2-rhodamine 110 (D2R), that was used to detect caspase activity in both apoptosis induced Jurkat and CEM cells. When apoptosis is induced, the caspases inside the cell cleave the D2R, creating free rhodamine 110. The fluorescence was measured using flow cytometry. The authors observed an increase in fluorescence within the cell over time in both cell types. In addition, when a caspase peptide inhibitor was added, no increase in fluorescence was observed. Although the authors used a probe with aspartic acid residues, it should be mentioned that these residues can and have been replaced with tetrapeptide sequences that are specific for a particular caspase. By using different caspase-selective peptide sequences, these fluorogenic probes can be used to identify different stages of caspase activity and therefore the temporal dynamics of apoptosis.Kohl et al. discovered a need for more sensitive measurements when investigating protease activity, specifically sensitive measurements within a single living cell. In their work in the Schwille group, they presented an in vivo protease assay that studied caspase reactions in situ using dual-color fluorescence cross-correlation spectroscopy (dcFCCS).32 The authors found that with substrate concentrations in the low nanomolar range, they could identify subtle changes in cellular processes. Although their results did not give any apoptosis information, it set the foundation for sensitive intracellular studies. Schwilles's group has also provided a step-by-step guide to applying fluorescence correlation spectroscopy to intracellular systems.33 In that work, the authors were able to generate correlation curves of fluorescently labeled cells using only one photon excitation. Based on their work, others have since applied FCS to apoptosis investigations.
Saito et al. used this foundation and investigated caspase-3 activity in vivo using dcFCCS.34 The authors did so by using chimeric proteins they constructed with fused enhanced green fluorescent protein (EGFP), and monomeric red FP (mRFP). In a control experiment, the authors were able to monitor the protease activity in solution. They observed a decrease in cross-correlation amplitude. When apoptosis was induced in a live cell, the authors were able to obtain consistent results by detecting a decrease in cross-correlation amplitude due to caspase-3 activation and cleavage of a specific amino-acid sequence. The authors give a clear example of the capability of FCCS to obtain sensitive results in detecting protease activity.
Recently, our group investigated caspase activity by fluorescence correlation spectroscopy (FCS).35 We were able to identify apoptotic Jurkat cells by the presence of the cleaved fluorescent probe D2R. In our studies, FCS curves were shown to be drastically different for autofluorescent (non-apoptotic) cells, and cells that cleaved the probe showed diffusion times and molecular brightness values characteristic of free rhodamine-110 (Fig. 2). The authors identified apoptotic cells as early as 45 min after induction with anti-CD95 based on molecular brightness (η), and molecular dwell time (τD). This method has shown several advantages over conventional methods due to high temporal resolution, and the ability to leave cells viable for further analysis. In addition, this method does not require transfection of the cell as in the case of previous studies. With these advantages, FCS has the capability of being a important tool for apoptosis studies.
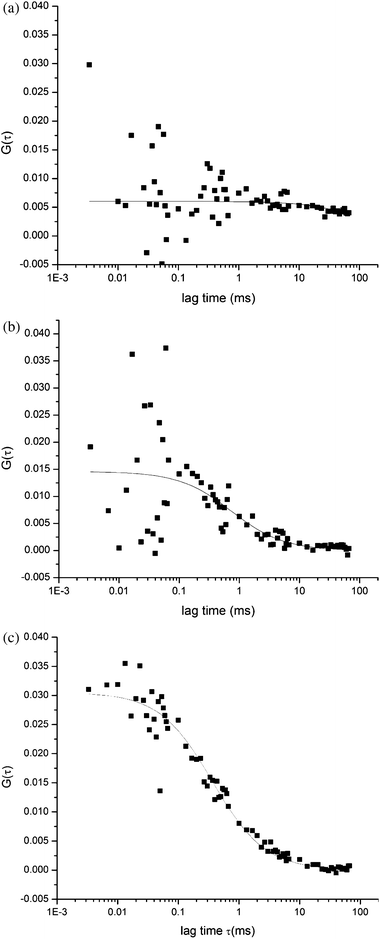 | ||
| Fig. 2 Fluorescence correlation spectroscopy measurements of apoptotic cells. A fluorogenic probe was detected inside single living cells. Autocorrelation functions for (a) a blank Jurkat cell, and (b) a control Jurkat cell, and (c) an induced cell after 45 min show markedly different levels of autocorrelation. The autocorrelation curve arises from the presence of rhodamine 110 after the fluorogenic substrate is cleaved. Reproduced with permission from Ref. 34. | ||
There is significant interest in the use of new fluorescent probes specific to one or more caspases. In the work done by Cai et al., new rhodamine 110 (R110) derivatives were synthesized to create a new dye and a new substrate that will increase the throughput of apoptotic cell detection.36 Unlike the traditional R110 based substrates that are bis-peptide-R110 derivatives, their approach uses one of the amino groups to introduce a blocking group as an enhancer moiety. This increases membrane penetration, therefore, increases substrate accumulation, and increases the retention of the fluorescent moiety inside the cell. They then used the other amino group to prepare a new substrate. Cells that were incubated with the dye, N-octyloxycarbonly-R110 (1), were highly fluorescent and were retained inside the cell longer than R110. The caspase-3 substrate, N-Ac-DEVD-N′-octyloxycarbonyl-R110, was tested using human recombinant caspase-3 (hrCasp3) and gave a strong fluorescent signal, while incubation with buffer gave a very low background signal. In addition, the substrate was also tested with lysates from HL-60 cells that were induced with an apoptotic-inducing agent. Again, when compared to an inhibitor (Ac-DEVD-CHO) there was strong fluorescent signal with the substrate, and almost no signal with the inhibitor.
In comparison with the fluorescence assay, Su et al. used an assay termed SAMDI-MS (self-assembled monolayers for matrix assisted laser desorption ionization time-of-flight mass spectrometry) to study caspase activity and apoptosis.37 In their assay, they determined caspase activity using monolayers specific for caspase-3 or caspase-8. The monolayers were exposed to lysates from Jurkat cells that were induced with staurosporin. The authors were able to detect caspase activity, label free, and without the loss of activity of the enzymes. In addition, by using longer peptide substrates, the assay proved to have better resolution than fluorescence assays.
There have also been recent advancements in biosensors to detect caspase activity. Specifically, in the work done by the Vo-Dinh group, an optical sensor having a nanoprobe was used to monitor the onset of caspase-9 activity by inducing the mitochondrial pathway of apoptosis.38 The Vo-Dinh group demonstrated the use of nanoprobes for the detection of caspase-9 activity in a single cell. This method is of increasing interest due to the minimally invasive nature of the probe. The nanoprobes allow penetration and sampling into a live cell without disrupting the cells normal functions. The authors use a fluorogenic enzyme substrate, Leucine-Glutamic Acid-Histidine-Aspartic Acid-7-amino-4-methylcourmarin (LEHD-AMC). In their experiment, human breast cancer cells (MCF-7 cells) were induced using photodynamic therapy protocols (PDT) with δ-aminolevulinic acid (ALA). The substrate tetrapeptide (LEHD-AMC) is cleaved by caspase 9 between aspartic acid and AMC. The fluorescence signal emitted by AMC is measured upon its excitation by a HeCd laser. The experiment was done in vitro using cell lysates and in vivo within single MCF-7 cells.
In a later work, Kihara et al. also implemented the use of a nanoprobe by coupling a nanoneedle on a cantilever and a fluorescent probe.39 This method, called Molecular Meter with Nanoneedle Technology (MOMENT), has been shown to be a minimally invasive procedure that utilizes atomic force microscopy (AFM) coupled with a fluorescent probe. MOMENT methods have the ability to analyze caspase activity and the potential to monitor other cellular processes. In their work, caspase-3 activity was observed using fluorescent resonance energy transfer (FRET) of their probe substrate (NHGcas546), which contains an Alexa Fluor 546 at its C-terminus. This probe was attached to the surface of a silicon cantilever, and its change in FRET signal was concluded as a result of cleavage by recombinant caspase-3. The same results occurred when the nanoneedle was inserted into an apoptotic cell. The fluorescence ratio decreased, and did not in normal cells.
4.6 Microfluidic applications
There has been increased interest in using microfluidic devices to study apoptosis. The majority of such devices examines caspase activity outside of cells or performs on-chip flow cytometry. One device demonstrated by Valero et al. was made of traps with various geometries designed to capture and hold cells in place.40 Once trapped, fluorescence imaging was used to monitor apoptosis induction in HL60 cells. In this case, induction was achieved using tumor necrosis factor-α (TNF-α). The sidewall traps enabled long-term observation of the cells as they progressed through the apoptotic process. Other traps and culture systems designed by Wlodkowic and co-workers41,42 have been used to screen for apoptosis-inducing drugs. Another device designed by Wu and co-workers has been demonstrated as an effective method of screening free caspase activity and inhibition.43 The device uses a split-injection method to mix a sample of caspases, a probe, and a substrate before detection downstream. Other microfluidic systems for apoptosis analysis have included electrophoresis chips for glutathione and hydrogen peroxide detection,44 sorting of mitochondria,45 and single-cell analysis of Cytochrome c distribution.46Another microfluidic device, demonstrated by Reif et al. in our group, used affinity interactions to capture Jurkat cells on a glass surface. In this case, the microfluidic channel was coated with anti-CD95, which resulted in the simultaneous capture and induction of the cells to undergo apoptosis. Annexin V-PE staining showed that 44% of the cells captured were apoptotic 3 h after capture on the channel surface (Fig. 3) relative to 8% by flow cytometry 3 h after induction. The use of this device provides precise information on the timing of apoptosis induction and allows for long term monitoring of captured cells. Devices such as those mentioned here have the potential to provide unique methods of studying various aspects of the apoptotic process.47
 | ||
| Fig. 3 White light and fluorescence images (×10 magnification) of Jurkat cells stained with Annexin V-PE and 7-AAD on the surface of an anti-CD95 coated channel 3.5 h after induction and binding. (a) white light image. (b) Annexin V-PE fluorescence. (c) 7-AAD fluorescence. The channel surface is coated with anti-CD95 antibodies that—upon binding—initiate apoptosis. Scale bar represents 100 μm. Reproduced with permission from Ref. 41. | ||
In a more recent work by Reif and co-workers in our laboratory, the same microdevice was applied to study the temporal dynamics of apoptosis.48 In this case, the cells were bound to the anti-CD95 surface of the device and then continuously stained with the caspase probe (D2R). The fluorescence intensity of the cells was monitored by light microscopy over a period of 6 h. Both the number of apoptotic cells over time and the fluorescence of individual cells were monitored simultaneously. The fluorescence intensity of individual cells was used to monitor the timing of caspase activity and the rate of D2R cleavage in each cell. The rate of caspase cleavage of D2R showed good agreement within the population. Interestingly, the time between induction and onset of caspase activity did not vary along with caspase cleavage rate. In addition, the caspase inhibitor, z-VAD-FMK, was shown to slow caspase activation and the D2R cleavage process. In the future, other caspase probes could also be used to further monitor the temporal dynamics of apoptosis.
4.7 Electrochemical methods
Methods to detect apoptosis do not all require complex and expensive instrumentation. Electrochemical methods have recently been presented as an alternative method to detect apoptotic cells. One of the early electrochemical sensors was presented by Xiao et al.49 The electrochemical sensor that they designed uses a gold electrode modified with a helix peptide ferrocene (Fc)-GDGDEVDGC. In this case, Fc is used as the electroactive reporter while the peptide sequence serves as a recognition and cleavage site of caspase 3. The general scheme for electrochemical detection of apoptosis is shown in Fig. 4. Using a modified electrode for cyclic voltammetry, active caspase 3 was detected in cell lysates based on the cleavage of the peptide sequence, which resulted in the loss of the reporter, Fc. In this way, the decrease in the peak current was a result of caspase 3 activity and could therefore be used to detect apoptosis. The signal decline was measured to be up to 85% in cell lysates of apoptosis induced cells with active caspase 3. This method of detecting apoptosis is simple and convenient as it does not require complicated instrumentation, only a modified electrode that is easy to create.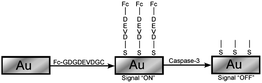 | ||
| Fig. 4 Schematic diagram for electrochemical detection. Cleavage of the DEVD peptide sequence releases the ferrocene (Fc) group, resulting in a decrease in the electrochemical signal. Electrochemical probes based on caspase cleavage allow label-free detection of caspase activity, and could be adapted for intracellular use using ultra-micro electrodes. Redrawn from Ref. 43. | ||
Other electrochemical methods use electrodes designed to detect the interaction between Annexin V and PS residues. An example is the recent design and demonstration of one such electrode presented by Liu and coworkers.50 The electrode is a pyrolytic graphite electrode co-modified with poly(ethylenimine) and Annexin V. Annexin V in the poly(ethylenimine) film is shown to retain its affinity for phosphatidylserine molecules, which are translocated from the inner to outer cell membrane during apoptosis. The binding of cells, in the presence of calcium, to the Annexin V in the electrode inhibits the redox reaction of [Ru(NH3)5Cl]2+/+ at the electrode surface. Changes in the shape of the cyclic voltammogram indicated the presence of apoptotic cells. The method has been shown to be a rapid and convenient way to detect apoptosis in cell solutions.50
Another electrochemical based biosensor developed by Tong et al. uses electrochemical impedance to detect the interaction between Annexin V and phosphatidylserine molecules externalized by apoptotic cells. The device featured high loading of Annexin V immobilized on a layer of self-assembled gold nanoparticles. The interaction between apoptotic cells and the Annexin V on the electrode surface was monitored by electrochemical impedance spectroscopy. The performance of the biosensor was validated using both a model system of cells integrated by phosphatidylserine-modified liposomes and a real sample of cells induced to undergo apoptosis using 5-fluorouracil. The results from the sensor matched those of two standard apoptosis detection techniques, flow cytometry and fluorescence microscopy. The method shows great promise for high throughput screening, medical filtration, and trace screening because of its cost-efficiency and simplicity.51
Electrochemical devices to detect apoptosis represent a new and evolving field of research. The major advantages of electrochemical methods include their throughput and ease of use. In addition, they are cost effective and simple to produce relative to many spectroscopic and flow cytometry methods. Their overall weakness is their inability to measure apoptosis on the single cell level or provide cell-specific information over a large cell population.
5. Conclusion
Apoptosis is an ever-growing field of study. The roles that apoptosis plays in human development and diseases, such as heart disease and cancer, make it crucial to understand the mechanisms of the process. Depending on the experiment, different methods are more useful than others. If, for example, one is interested in the morphology of apoptotic cells, electron microscopy is probably the most informative method of detecting apoptosis, while fluorescence (and white light) microscopy can also be used. If on the other hand, measuring large numbers of cells is required, flow cytometry is likely the best method of detection. If the cells need to be monitored over long periods of time, light microscopy and microfluidic devices provide an easy and versatile method to do so. The presence of proteins in apoptotic cells can be monitored by western blotting, while if DNA is the object of study, the DNA ladder assay can provide information of the extent of DNA degradation. The common methods and their sensitivities are summarized in Table 1.| Method | Detection Time | Number of Cells | Cells Viable? | Sensitivity |
|---|---|---|---|---|
| a Not recoverable. | ||||
| Fluorescence Correlation Spectroscopy | 30–45 Minutes | Single | Yes | Single Molecule (<pM) |
| Electron Microscopy | 16–60 Minutes | Single | No | N A−1 |
| Western Blotting | 30 Minutes | Cell Lysates | No | pM-nM |
| Annexin V Staining | 1–2 Hours | Single or Many | Yes | nM |
| TUNEL Assay | 2 Hours | Single or Many | No | N A−1 |
| Flow Cytometry | 2 Hours | Several | Yesa | pM-nM |
Despite the many methods of apoptosis detection and monitoring, there are still many challenges. Optimization and improvements of the spatial/temporal resolution, sensitivity, and ease of use of current approaches remains an ongoing effort. The discovery of new methods will also undoubtedly change the way apoptosis is studied both in culture and in tissue. Other points of analyses, such as the detection of regulatory proteins in intact cells, will change the way apoptosis is detected entirely. New analytical targets, and analyses targeting multiple steps in the process, must be continually introduced and improved so that, ultimately, the mechanisms of apoptosis can be unravelled and understood.
Acknowledgements
Michelle M. Martinez was supported by a Provost Fellowship from Texas Tech University. Randall D. Reif and Dimitri Pappas were supported by a grant from the Robert A. Welch Foundation (Grant D-1667).References
- Y. Otsuki, L. Zhonglian and S. Masa-Ai, Prog. Histochem. Cytochem., 2003, 38, 275–340 CrossRef.
- H. Yaoita, K. Ogawa, K. Maehara and Y. Maruyama, Circulation, 1998, 97, 276–281 CAS.
- B. Fadeel and S. Orrenius, J. Intern. Med., 2005, 258, 479–517 CrossRef CAS.
- T. A. Holly, A. Drincic, Y. Byun, S. Nakamura, K. Harris, F. J. Klocke and V. L. Cryns, J. Mol. Cell. Cardiol., 1999, 31, 1709–1715 CrossRef CAS.
- M. A. O'Brien and R. Kirby, Journal of Veterinary Emergency and Critical Care, 2008, 18, 572–585 CrossRef.
- R. S. Hotchkiss, K. W. Tinsley and I. E. Karl, J Infect Dis, 2003, 35, 585–592 CAS.
- I. M. Ghorial, T. E. Witzig and A. A. Adjei, Ca–Cancer J. Clin., 2005, 55, 178–194 Search PubMed.
- E. Finkel, Science, 2001, 292, 624–626 CrossRef CAS.
- J. G. Tidball, D. E. Albrecht, B. E. Lokensgard and M. J. Spencer, Journal of Cell Science, 1995, 108, 2197–2204 CAS.
- Y. Lee and A. B. Gustafsson, Apoptosis, 2009, 14, 536–548 Search PubMed.
- S. Elmore, Toxicol. Pathol., 2007, 35, 495–516 CrossRef CAS.
- G. M. Salido and J. A. Rosado (ed.), Apoptosis: Involvement of Oxidative Stress and Intracellular Ca2+ Homeostasis, Springer 2009 Search PubMed.
- D. Pappas, Practical Cell Analysis, Wiley 2010 Search PubMed.
- S. Yasuhara, Y. Zhu, T. Matsui, N. Tipirneni, Y. Yasuhara, M. Kaneki, A. Rosenzweig and J. A. J. Martyn, The Journal of Hisochemistry&Cytochemistry., 2003, 51, 873–885 Search PubMed.
- K. Roberg, Laboratory Investigation, 2001, 81, 149–158 CAS.
- A. Boubouti, P. T. Doulias, L. Nousis, M. Tenopoulou and D. Galaris, Free Radical Biol. Med., 2002, 33, 691–702 CrossRef.
- G. Koopman, C. P. Reutelingsperger, G. A. Kuijten, R. M. Kechnen, S. T. Pals and M. H. van Oers, Blood, 1994, 8, 1415–1420.
- M. van England, L. J. W. Nieland, F. C. S. Ramackers, B. Schutte and C. P. M. Reutelingsperger, Cytometry, 1998, 31, 1–9 CrossRef CAS.
- R. Gatti, S. Belletti, G. Orlandini, O. Bussolati, V. Dall'Asta and G. C. Gazzola, The Journal of Hisochemistry&Cytochemistry, 1998, 46, 895–900 Search PubMed.
- O. Bussolati, S. Bellettti, J. Uggeri, R. Gatti, G. Orlandini, V. Dall' Asta and G. C. Gazzola, Exp. Cell Res., 1995, 220, 283–291 CrossRef CAS.
- Y. Gavrieli, Y. Sherman and S. A. Ben-Sasson, J. Cell Biol., 1992, 119, 493–501 CrossRef.
- S. K. Ray, K. E. Schaecher, D. C. Shields, E. L. Hogan and N. L. Banik, Brain Res. Protoc., 2000, 5, 305–311 CrossRef CAS.
- T. Imasawa, K. Koike, I. Ishii, J. Chun and Y. Yatomi, Biochem. Biophys. Res. Commun., 2010, 392, 207–211 CrossRef CAS.
- E. Jacotot, L. Ravagnan, M. Loeffler, K. F. Ferri, H. L. Vieira, N. Zamzami, P. Costantini, S. Druillennec, J. Hoebeke, J. P. Briand, T. Irinopoulou, E. Daugas, S. A. Susin, D. Cointe, Z. H. Xie, J. C. Reed, B. P. Roques and G. Kroemer, J. Exp. Med., 2000, 191, 33–46 CrossRef CAS.
- C. Ferlini, S. Di Cesare, G. Rainaldi, W. Malorni, P. Samoggia, R. Biselli and A. Fattorossi, Cytometry, 1996, 24, 106–115 CrossRef CAS.
- M. Poot, L. L. Gibson and V. L. Singer, Cytometry, 1997, 27, 358–364 CrossRef CAS.
- J. Dobrucki and Z. Darzynkiweicz, Micron, 2001, 32, 645–652 CrossRef CAS.
- U. S. Babu, D. M. Gaines, Y. Wu, C. D. Westphal, M. Pereira and R. B. Raybourne, J. Microbiol. Methods, 2008, 75, 167–171 CrossRef CAS.
- D. Wlodkowic, J. Skommer, S. Faley, Z. Darzynkiewicz and J. M. Cooper, Exp. Cell Res., 2009, 315, 1706–1714 CrossRef CAS.
- H. R. Stennicke, C. A. Ryan and G. S. Salvesen, Trends Biochem. Sci., 2002, 27, 94–98 CrossRef CAS.
- H. Hug, M. Los, H. Werner and K. Debatin, Biochemistry, 1999, 38, 13906–13911 CrossRef CAS.
- T. Kohl, E. Haustein and P. Schwille, Biophys. J., 2005, 89, 2770–2782 CrossRef CAS.
- S. A. Kim, K. G. Heinze and P. Schwille, Nat. Methods, 2007, 4, 963–973 CrossRef CAS.
- K. Saito, I. Wada, M. Tamura and M. Kinjo, Biochem. Biophys. Res. Commun., 2004, 324, 849–854 CrossRef CAS.
- M. M. Martinez, R. D. Reif and D. Pappas, Anal. Bioanal. Chem., 2010, 396, 1177–1185 CrossRef CAS.
- S. X. Cai, H. Zhang, J. Guastella, J. Drewe, W. Yang and E. Weber, Bioorg. Med. Chem. Lett., 2001, 11, 39–42 CrossRef CAS.
- J. Su, T. W. Rajapaksha, M. E. Peter and M. Mrksich, Anal. Chem., 2006, 78, 4945–4951 CrossRef CAS.
- P. M. Kasili, J. M. Song and T. Vo-Dinh, J. Am. Chem. Soc., 2004, 126, 2799–2806 CrossRef CAS.
- T. Kihara, C. Nakamura, M. Suzuki, S. Han, K. Fukazawa, K. Ishihara and J. Miyake, Biosens. Bioelectron., 2009, 25, 22–27 CrossRef CAS.
- A. Valero, F. Merino, F. Wolbers, R. Luttge, I. Vermes, H. Andersson and A. ven den Berg, Lab Chip, 2005, 5, 49–55 RSC.
- D. Wlodkowic, S. Faley, M. Zagnoni, J. P. Wikswo and J. M. Cooper, Anal. Chem., 2009, 81, 5517–5523 CrossRef CAS.
- D. Wlodkowic, J. Skommer, D. McGuinness, S. Faley, W. Kolch, Z. Darzynkiewicz and J. M. Cooper, Anal. Chem., 2009, 81, 6952–6959 CrossRef CAS.
- G. Wu, J. Irvine, C. Luft, D. Pressley, C. N. Hodge and B. Janzen, Combinatorial and High Throughput Screening, 2003, 6, 303–312 Search PubMed.
- Z. Chen, Q. Li, X. Wang, Z. Wang, R. Zhang, M. Yin, L. Yin, K. Xu and B. Tang, Anal. Chem., 2010, 82, 2006–2012 CrossRef CAS.
- H. Lu, S. Gaudet, M. A. Schmidt and K. F. Jensen, Anal. Chem., 2004, 76, 5705–5712 CrossRef CAS.
- E. Tamaki, K. Sato, M. Tokeshi, K. Sato, M. Aihara and T. Kitamori, Anal. Chem., 2002, 74, 1560–1564 CrossRef CAS.
- R. D. Reif, M. M. Martinez, K. Wang and D. Pappas, Anal. Bioanal. Chem., 2009, 395, 787–795 CrossRef CAS.
- R. D. Reif, C. Aguas, M. M. M. Martinez and D. Pappas, Anal. Bioanal. Chem., 2010 DOI:10.1007/s00216-010-3567-1.
- H. Xiao, L. Liu, F. Meng, J. Huang and G. Li, Anal. Chem., 2008, 80, 5272–5276 CrossRef CAS.
- T. Liu, W. Zhu, X. Yang, L. Chen, R. Yang, Z. Hus and G. Li, Anal. Chem., 2009, 81, 2410–2413 CrossRef CAS.
- C. Tong, B. Shi, X. Xiao, H. Liao, Y. Zheng, G. Shen, D. Tang and X. Liu, Biosens. Bioelectron., 2009, 24, 1777–1782 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |