Enhancing determination of organophosphate species in high inorganic phosphate matrices: application to nerve agent degradation products
Yaofang
Zhang
a,
Kevin M.
Kubachka
ab and
Joseph A.
Caruso
*a
aDepartment of Chemistry, University of Cincinnati, Cincinnati, OH 45221, USA. E-mail: joseph.caruso@uc.edu
bForensic Chemistry Center, United States Food and Drug Administration, Cincinnati, OH 45237, USA
First published on 3rd August 2010
Abstract
Developing a reliable and robust trace level method for organophosphates in the presence of relatively high inorganic phosphate is an area of interest for further method development. Since the advent of collision/reaction cell technology, inductively coupled plasma mass spectrometry (ICPMS) has been used with a variety of sample types for elemental phosphorus detection at m/z = 31. However, with many samples inorganic phosphate may be orders of magnitude higher than organophosphates, presenting a major interference to quantification. Therefore, removal of inorganic phosphate to levels low enough to minimize the interference would prove beneficial for more effective organophosphate analyses. In this study, applicable to most organophosphate containing samples, the illustration is to nerve agent degradation products as they might contaminate food and environmental systems after their initial formation. Calcium chloride was chosen as a coagulant mix and ammonium hydroxide was chosen to adjust the pH in order to remove inorganic phosphate at the sample preparation step. High performance liquid chromatography (HPLC) coupled with ICPMS was utilized for analysis. The results show that inorganic phosphate, at concentrations as high as 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 µg mL−1, can be sufficiently removed by precipitation with calcium at pH > 9 while the organophosphorus chemical warfare agent degradation products, CWADPs, remain intact. Applications to apple juice and cola drink indicate that this method is suitable for more complex matrices containing relatively high levels of inorganic phosphate.
000 µg mL−1, can be sufficiently removed by precipitation with calcium at pH > 9 while the organophosphorus chemical warfare agent degradation products, CWADPs, remain intact. Applications to apple juice and cola drink indicate that this method is suitable for more complex matrices containing relatively high levels of inorganic phosphate.
Introduction
Trace level organophosphates in the presence of relatively high concentrations of inorganic phosphate are an area of interest for further method development because of large inorganic phosphate interferences common to real-world sample matrices. Since the advent of collision/reaction cell technology, quadrupole ICPMS has the capability to analyze part-per-billion levels of phosphorus. The detection is viam/z = 31 at the only natural isotope of phosphorus 31P or at m/z = 47, the PO+ moiety with quadrupole detection for both. High resolution sector-field ICPMS may also be used. With the first method, the 15NO+ and NOH+ have to be removed or treated so they do not reach the quadrupole, since these are the major interferences at m/z = 31. In the second instance, PO+ is formed, thereby removing the phosphorus from the m/z = 31 interferences. And lastly the high resolution instrument separates 31P+ from the predominant interferences via spectral resolution.Elemental phosphorus detection has been used with a variety of sample types such as human plasma, fire retardants, pesticides, herbicides, nerve agent degradation products (NADPs), foods, beverages, etc.1–4 However, with many samples, inorganic phosphate may be orders of magnitude higher than organophosphates, presenting a major interference to quantification. In these determinations both liquid and gas chromatography have been found highly useful as the separation with ICPMS detection.
In view of the structural similarities of the low molecular weight nerve agent degradation products to pesticides, fire retardants, certain plasticizers, herbicides and other organophosphorus compounds in common use, these were chosen to illustrate the general applicability of this particular method. Additionally, possible contamination of the foods supply with these NADPs is a national concern. With many food and beverages containing inorganic phosphate, developing trace level organophosphate methods that are relatively interference free at ppb levels or below is timely and appropriate.
Various techniques have been used to analyze organophosphates.5–10 The most popular and traditional screening methods are GC and HPLC with specific detectors (FPD, NPD for GC and UV for LC) and selected spectrometries (MS or FTIR).9GC separation is more suitable for volatile and reactive analytes such as parent nerve agents (NAs). And HPLC is more useful for less volatile, more polar/water soluble molecules such as NADPs. In addition to conventional chromatographic separation techniques, capillary electrophoresis (CE) is also an alternative choice with high efficiency and relatively short analysis times.11,12 The techniques for identification and detection have been utilized and developed with MS, FTIR and NMR, but the most predominate one is chromatography coupled to MS.13–18 However, there may be deleterious effects from the ionization step leading to relatively higher detection limits.5,9 Thus, ICPMS using 31P+ as the target element may be most useful because of its high ionization capability, the ability to preclude the NOH+ and 15NO+ interferences with collision/reaction cells, and convenient interface towards various separation techniques such as GC, LC and CE.19–23
Among those chemical warfare agents, nerve agents are one of the most toxic and predominant family posing a deadly threat upon accidental or intentional release. There are two classes of nerve agents: G-type (more volatile, less persistent) and V-type (less volatile, more persistent). G-Type agents include: tabun (GA), sarin (GB), soman (GD), cyclosarin (GE and GF) and V-type agent comprises VE, VG, VM, VX, and r-VX. Upon introduction to the environment, they hydrolyze forming their respective degradation products.
Kubachka et al. developed a robust separation method, capable of separating 10 NADPs in the presence of inorganic phosphate with applications involving a wide range of food matrices.24 However, at concentrations of inorganic phosphate >1000 µg mL−1, phosphate could not be resolved from methylphosphonic acid (MPA) and ethylphosphonic acid (EPA) and the excessive peak tailing interfered with later eluting NADPs, as well. According to hydrolysis pathways of nerve agents,25,26 many eventually yield MPA, as a final degradation product, while VE can degrade to ethylphosphonic acid, EPA. Hence, MPA is a good indicator of previous nerve agents, and EPA plus MPA together suggest the earlier presence of VE, however, these compounds are difficult to detect in the presence of an high inorganic phosphate interference. Thus, the approach taken here is to remove the inorganic phosphate to the level where its interference may be compensated for and a viable analysis achieved.
In this study, a rapid, robust and simple approach for sample preparation was explored to reduce the inorganic phosphate interference while preserving organophosphates through chemical precipitation. The anion exchange LC-ICPMS method was employed for separation and detection after sample treatment. This method was applied to various sample matrices and was able to reduce or remove the inorganic phosphate for better detection ofNADPs.
Calcium chloride and ammonium hydroxide were chosen as mixes to adjust the pH and to remove inorganic phosphate at the sample preparation step. HPLC was utilized with ICPMS detection. Applications to apple juice and cola drink indicate that this method is suitable for more complex matrices containing relatively high levels of inorganic phosphate.
Experimental
Reagents and materials
All water was doubly distilled and deionized (DDW) by passing through a Millipore (18 MΩ) filtration system (Bedford, MA). The reagents utilized throughout these experiments were analytical grade or higher and without further purification. Formic acid (FA) and calcium chloride were purchased from Fisher Scientific (Fair Lawn, NJ). Methanol was acquired from Tedia (Fairfield, OH). Nitric acid, ammoniumhydrogen phosphate ((NH4)2HPO4) and ammonium carbonate ((NH4)2CO3) were supplied by J. T. Baker (Phillipsburg, NJ). Ammonium hydroxide (29.8% NH4OH, w/w) was obtained from Pharmco (Brookfield, CT). The solutions of 2.5% (v/v) formic acid with 5% (v/v) methanol in DDW, 0.5% (v/v) formic acid with 5% (v/v) methanol in DDW and 0.3 M NH4HCO2 with 22% MeOH in DDW were used as chromatographic buffers. Those buffers were prepared fresh from stock solution before starting the experiments.Food samples were purchased at local markets.
Standards
The chemical warfare degradation products, methylphosphonic acid (MPA, 2), ethylphosphonic acid (EPA, 4), propylphosphonic acid (PPA, 6), were obtained from Sigma-Aldrich in powder form, and were prepared as 5000 µg mL−1 standards in MeOH. Stock solutions of ethylhydrogen dimethylaminophosphate, sodium salt (EHDAP, 1), dimethylhydrogen phosphate (DMHP, 5), ethyl methylphosphonic acid (EMPA, 7), isopropyl methylphosphonic acid (IMPA, 8), diethyl hydrogen phosphate (DEHP, 9), and isopropylhydrogen ethylphosphonate (IPHEP, 10), in MeOH at 1000 µg mL−1 of each, were purchased from Cerilliant (Round Rock, TX). Abbreviations of CWADPs vary among different publications; abbreviations in this work were partially adopted from Black and Read.27 The structures of NADPs in this study have been demonstrated in previous literature.24–26 All standards of CWADPs were kept in amber vials at −20 °C. Stock solutions were diluted with DDW to appropriate concentrations prior to analysis.Instrumentation
Result and discussion
Choice of precipitant and pH on precipitation of inorganic phosphate
The acid dissociation constants for inorganic phosphorous acid are pKa1 = 2.12, pKa2 = 7.2 and pKa3 = 12.3. Depending on the nature of the sample, they may contain various forms of inorganic phosphate (H3PO4, H2PO4−, HPO42−, or PO43−). Only monohydrogen phosphate HPO42− and orthophosphate PO43− can form precipitates with the selected cations. Therefore, pH is an important parameter to assure the formation of these two species for removal of inorganic phosphate. In wastewater, the pH value is increased beyond about 10 to achieve high precipitation levels. To minimize the introduction of low ionization potential cations such as Na and K, which could be deleterious to the sensitivity for phosphorus, NH4OH was chosen as a volatile base for sample pH adjustment. When choosing the precipitant, it was noted that many of the common metal cations, Ca, Fe, Al, and Mg also form precipitates with hydroxide. Thus, hydroxideprecipitation is an inherent competitor to phosphateprecipitation. A lower Ksp of phosphate precipitate with a higher Ksp of hydroxide precipitate favors the phosphateprecipitation for the given metal cation. When examining the Ksp of the metal cations with HPO42−/PO43− and OH−, Ca exhibited a large difference in Ksp28 and was chosen as the cation for phosphateprecipitation.Studies were then performed to optimize the pH and the amount of CaCl2. To optimize the pH, CaCl2 was added in excess to a phosphate solution, the pH was adjusted to various values and the supernatant was tested for relative 31P signal using ICPMS. A significant decrease in signal was noted at pH > 9 (data not shown). The mole ratio of Ca:P for the treatment should be greater than 1:1, given the formula of the precipitates are Ca(HPO4)2 and CaPO4 and also that some of the Ca is likely consumed by precipitation with OH−.
Treatments on standard mix samples and HPLC-ICPMS detection
After choosing the phosphate precipitant and optimizing pH, three solutions with levels of inorganic phosphate HPO42− (100, 1000, 10000 µg mL−1), fortified with 5 µg mL−1MPA and EPA in DDW were prepared. A portion of the initial solutions was set aside for analysis by LC-ICPMS to be compared with the following treatments. For Treatment 1, the pH was adjusted with ammonia, to deprotonate basic phosphate to orthophosphate (pH > 9), then CaCl2 solution was introduced for precipitation. In summary, in Treatment 1, three mixtures were prepared – one with 0.02% NH4OH (v/v), 5 mM CaCl2, and 100 μg mL−1 HPO42− ; a second with 0.2% NH4OH, 50 mM CaCl2, and 1000 μg mL−1 HPO42− ; and a third with 0.2% NH4OH, 250 mM CaCl2, and 10000 μg mL−1 HPO42− – each containing 5 µg mL−1MPA and EPA. The supernatant was filtered using a 0.22 µm cellulose acetate centrifuge filter and the final filtrate was set aside for LC-ICPMS analysis. In Treatment 2, 200 µL final filtrate solution from Treatment 1 was added and spiked with 1 mM, 10 mM, 100 mM of (NH4)2CO3 (final concentration) for three inorganic phosphate levels, respectively. This step was included to remove excess calcium (via CaCO3precipitation, discussed later). This solution was then filtered through a 0.22 µm cellulose acetate centrifuge filter, and set aside for LC-ICPMS analysis. The pH values of final filtrates for the Treatment 1 and 2 trials were approximately 9. The three solutions (for each inorganic phosphate level) were introduced viaHPLC separation using an anion exchange column, the Hamilton PRP-X100, with 2.5% (v/v) formic acid and 5% (v/v) methanol in DDW as isocratic mobile phase, and the eluent was introduced to ICPMS for 31P specific detection. Additionally a standard mixture of HPO42−, MPA and EPA at 5 µg mL−1 as well as samples spike with the respective standards was analyzed to confirm peak identities.
As shown in Fig. 1, the EPA peak is overlapped by the HPO42− peak at 100 µg mL−1. As inorganic phosphate concentration increases from 100 to 1000 then to 10000 µg mL−1, the MPA and EPA peaks were both completely masked by HPO42− (data not shown). After Treatment 1, pH adjusted to >9 and addition of CaCl2, the HPO42− peak was dramatically reduced, while MPA and EPA were retained, with little peak overlap among the three compounds. Adding ammonium carbonate for the removal of excess calcium (Treatment 2) did not significantly affect the chromatogram compared to only Treatment 1 for HPO42− at 100 µg mL−1. When Treatment 2 was performed on the 1000 and 10000 µg mL−1 HPO42− solution mixtures, the area of the HPO42− peak virtually disappeared (Fig. 2(1) and (2)). Additionally, the area of the MPA and EPA peaks decreased slightly in the 1000 µg mL−1 HPO42− solution mixture (Fig. 2(1)). For the 10000 µg mL−1 HPO42− level, the MPA and EPA peaks decreased significantly after Treatment 1 (suppressed to 20% and 33% of peak areas for untreated MPA and EPA) and are reduced even further after Treatment 2 (decreased to 8% and 32% of peak areas for untreated MPA and EPA (Fig. 2(2))). This is probably due to the high amount of CaCl2 (250 mM) needed to precipitate phosphate at high levels. Relative to the MPA and EPA concentrations, this provides a significant amount of Ca3(PO4)2 precipitate and of excess calcium, resulting in more CaCO3 (as a result of Treatment 2) being formed, which might cause MPA and EPA to co-precipitate. Thus, Treatment 2 is unnecessary for samples with very high concentration of phosphate due to the reduced response toMPA and EPA.
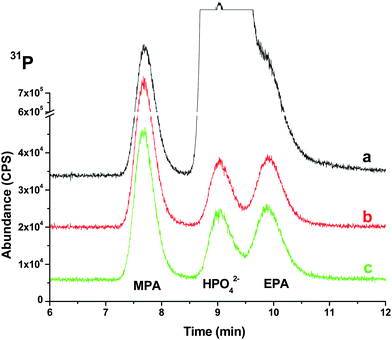 | ||
| Fig. 1 (a) 100 µg mL−1 HPO42− fortified with 5 µg mL−1MPA and EPA, (b) 100 µg mL−1 fortified HPO42− treated with NH4OH (0.2%) and CaCl2 (5mM) (Treatment 1) and (c) NH4OH (0.2%) and CaCl2 (5mM) treated filtrate added with (NH4)2CO3 (1mM) (Treatment 2). | ||
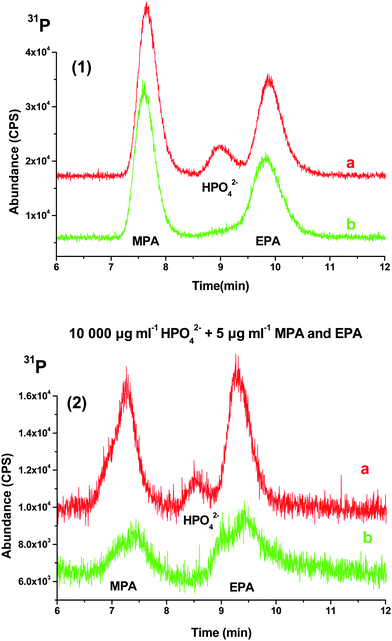 | ||
| Fig. 2 (1) and (2) represent 1000 µg mL−1 and 10000 µg mL−1 HPO42− respectively with 5 µg mL−1 of MPA and EPA fortified (a) result after NH4OH and CaCl2 treatment, and (b) result of NH4OH and CaCl2 treated filtrate added with (NH4)2CO3. | ||
Moreover, as the inorganic level increased, the retention time of MPA and EPA decreased after treatment with ammonia and CaCl2 as shown in Fig. 3. This is probably due to the competition between the Ca ion and the stationary phase of the column. The anion exchange column has styrene divinylbenzene substrate that has been derivatized chemically to introduce trimethylammonium quaternary sites with positive charges.29 As the amount of Ca2+ ion increased, more analytes might interact with Ca2+ ion instead of trimethylammonium quaternary site and therefore, eluted earlier. Thus, the higher level of inorganic phosphate required more CaCl2 causing more Ca2+ to remain after treatment and subsequently, a greater retention time shift.
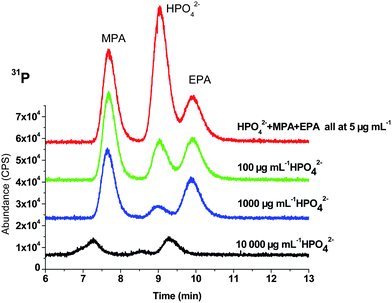 | ||
| Fig. 3 Comparison of results of three levels of HPO42− fortified with MPA and EPA after treated with NH4OH and CaCl2. | ||
Therefore, in a simple solution, water-based matrix, removal of inorganic phosphate interferences can be successfully achieved by calcium addition at pH > 9, with minimal effect on the closely eluting MPA and EPA peaks. The next step was to test the method performance for samples with more complex matrices and to examine the effect on additional CWADPs.
Application on real world samples with fortification of additional CWADPs
The preliminary research24,30 demonstrated that food samples contain higher concentration of inorganic phosphate and have more complex matrices than other environmental samples such as drinkingwater, ground water, and soil extracts. To examine the capability and practicability of this method for inorganic phosphate removal, several food samples were tested. The first step was to determine the total 31P concentrations (assuming all 31P to be from inorganic phosphate) of several food and beverage samples determined using ICPMS with external calibration: apple juice 130 µg mL−1, orange juice 330 µg mL−1, cola drink 270 µg mL−1.The sample preparation procedures discussed above were performed and, additionally, the samples were degassed by sonication to reduce dissolved carbon dioxide, which can interfere with the precipitation efficiency of calcium. Also treatment with (NH4)2CO3 (Treatment 2) was omitted. Additional NADPs (EHDAP, DMHP, PPA, EMPA, IMPA, DEHP and IPHEP) were fortified into the sample at concentrations of 5 µg mL−1 to examine the effect of precipitation on each of the NADPs. The separation method adapted from Kubachka et al. was used: mobile phase A and B (A: 0.5% (v/v) formic acid with 5% (v/v) methanol in DDW and B: 0.3 M NH4HCO2 (pH ≈ 2.3), 22% MeOH in DDW) at a flow rate of 1.0 mL min−1 with the following gradient : 0–10 min, 0% B; 10–12 min, 0–10% B; 12–15 min, 10% B; 15–18 min, 10–70% B; 18–24 min, 70% B; 24–29 min, 70–0% B.
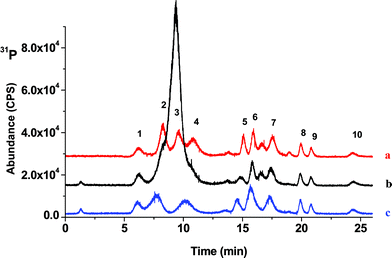
For preparation of the apple juice or cola samples, each was diluted 1:10 with DDW, then NH4OH and CaCl2 were added to a final concentration of 2% (v/v) and 10 mM, respectively. Fig. 4 shows the chromatograms of NADP standard mixtures with inorganic phosphate each at 5 µg mL−1, diluted apple juice (untreated), and apple juice spiked with 5 µg mL−1 of NADPs after Treatment 1. Fig. 5 represents similar conditions while examining cola drink rather than apple juice. As shown in Fig. 5, before treatment, the intrinsic inorganic phosphate in cola drink overlapped both MPA and EPA, making their detection or quantification difficult. Once Treatment 1 was carried out on the samples, inorganic phosphate was largely reduced to the level that the chromatographic peaks of MPA, EPA and HPO42− could be resolved. It is also important to note that no effect was shown on the separation and detection of the other CWADPs. The retention times of all compounds shifted slightly after Treatment 1, but not enough to complicate identifying the CWADPs plus the retention order remained the same.
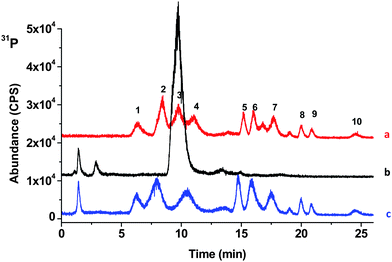 | ||
| Fig. 4 (a) HPO42− and NADP standards all at 5 µg mL−1, (b) diluted apple juice and (c) diluted apple juice fortified with 5 µg mL−1NADP standards after treatment with NH4OH (2%)and CaCl2 (10 mM) (pH ∼11). (1) EHDAP, (2) MPA, (3) H2PO4−, (4) EPA, (5) DMHP, (6) PPA, (7) EMPA, (8) IMPA, (9) DEHP and (10) IPHEP. | ||
 | ||
| Fig. 5 (a) HPO42− and NADP standards all at 5 µg mL−1, (b) diluted cola drink fortified with 5 µg mL−1NADP standards and (c) diluted cola drink fortified with 5 µg mL−1NADP standards after treatment with NH4OH (2%) and CaCl2 (10mM) (pH ∼11). | ||
Conclusion
In this study, a rapid and simple sample treatment method for reducing inorganic phosphate interference for previously developed chromatographic separations analyzing organophosphorus CWADPs was investigated. Ammonium hydroxide was added to adjust the pH > 9, then calcium chloride was chosen as a precipitant for inorganic phosphate. High concentrations of inorganic phosphate (as high as 10000 µg mL−1 (1% inorganic phosphate solution)) interference that overlapped MPA and EPA chromatographic peaks were reduced to the level that allowed the chromatographic resolution of inorganic phosphate from MPA and EPA. This treatment works for various concentration levels of inorganic phosphate from 100 µg mL−1 up to 10000 µg mL−1 while allowing 10 CWADPs to remain relatively phosphate interference free. Applications of the developed method to food samples including apple juice and cola drink demonstrated that phosphateprecipitation is effective for high level removal of inorganic phosphates. This method may prove useful in other mass spectrometric analysis approaches, such as LC-electrospray ionization MS, which will be intolerant of such complicated matrices.
Abbreviations
| EHDAP | Ethyl hydrogen dimethylaminophosphate, sodium salt |
| MPA | Methylphosphonic acid |
| EPA | Ethylphosphonic acid |
| DMHP | Dimethyl hydrogen phosphate |
| PPA | Propylphosphonic acid |
| EMPA | Ethyl methylphosphonic acid |
| IMPA | Isopropyl methylphosphonic acid |
| DEHP | Diethyl hydrogen phosphate |
| IPHEP | Isopropyl hydrogen ethylphosphonate |
Acknowledgements
YZ and JAC would like to thank NIEHS grant # ES04908 for partial funding this research. We also acknowledge Agilent Technologies for their continuing support of our studies at the University of Cincinnati/Agilent Technologies Metallomics Center of the Americas.Reference
- J. Ellis, et al., Determination of organophosphorus fire retardants and plasticizers in wastewater samples using MAE-SPME with GC-ICPMS and GC-TOFMS detection, J. Environ. Monit., 2007, 9(12), 1329–1336 RSC.
- B. B. M. Sadi, A. P. Vonderheide and J. A. Caruso, Analysis of phosphorus herbicides by ion-pairing reversed-phase liquid chromatography coupled to inductively coupled plasma mass spectrometry with octapole reaction cell, J. Chromatogr., A, 2004, 1050(1), 95–101 CrossRef CAS.
- M. Shah and J. A. Caruso, Inductively coupled plasma mass spectrometry in separation techniques: recent trends in phosphorus speciation, J. Sep. Sci., 2005, 28(15), 1969–1984 CrossRef CAS.
- M. Shah, et al., Determination of phosphoric acid triesters in human plasma using solid-phase microextraction and gas chromatography coupled to inductively coupled plasma mass spectrometry, J. Chromatogr., A, 2006, 1103(2), 329–336 CrossRef.
- P. A. D'Agostino and C. L. Chenier, Analysis of Chemical Warfare Agents: General Overview, LC-MS Review, In-House LC-ESI-MS Methods and Open Literature Bibliography, Defense Research and Development Canada, Suffield, 2006 Search PubMed.
- I. Ferrer and E. M. Thurman, Multi-residue method for the analysis of 101 pesticides and their degradates in food and water samples by liquid chromatography/time-of-flight mass spectrometry, J. Chromatogr., A, 2007, 1175(1), 24–37 CrossRef CAS.
- M. Garcia, I. Rodriguez and R. Cela, Microwave-assisted extraction of organophosphate flame retardants and plasticizers from indoor dust samples, J. Chromatogr., A, 2007, 1152(1–2), 280–286 CrossRef CAS.
- H. H. Hill, Jr and S. J. Martin, Conventional analytical methods for chemical warfare agents, Pure Appl. Chem., 2002, 74(12), 2281–2291 CrossRef.
- M. Mesilaakso, Chemical Weapons Convention Chemicals Analysis: Sample Collection, Preparation and Analytical Methods, Wiley, Chichester, 2005 Search PubMed.
- K. Y. O. Yin Sun, Detection Technologies for Chemical Warfare Agents and Toxic Vapors, CRC Press, 2005 Search PubMed.
- J. P. Mercier, et al., Identification of phosphonic acids by capillary electrophoresis-ionspray mass spectrometry, J. Chromatogr., A, 1998, 825(1), 71–80 CrossRef CAS.
- A. F. Nassar, S. V. Lucas and L. D. Hoffland, Determination of chemical warfare agent degradation products at low-part-per-billion levels in aqueous samples and sub-part-per-million levels in soils using capillary electrophoresis, Anal. Chem., 1999, 71(7), 1285–1292 CrossRef CAS.
- R. M. Black and R. W. Read, Analysis of degradation products of organophosphorus chemical warfare agents and related compounds by liquid chromatography-mass spectrometry using electrospray and atmospheric pressure chemical ionisation, J. Chromatogr., A, 1998, 794(1–2), 233–244 CrossRef CAS.
- G. L. Hook, et al., Detection of VX contamination in soil through solid-phase microextraction sampling and gas chromatography/mass spectrometry of the VX degradation product bis(diisopropylaminoethyl)disulfide, J. Chromatogr., A, 2003, 992(1–2), 1–9 CrossRef CAS.
- C. E. Kientz, Chromatography and mass spectrometry of chemical warfare agents, toxins and related compounds: state of the art and future prospects, J. Chromatogr., A, 1998, 814(1–2), 1–23 CrossRef CAS.
- M. L. Li, et al., Identification of certain chemical agents in complex organic solutions by gas chromatography/tandem mass spectrometry, J. Mass Spectrom., 2006, 41(11), 1453–1458 CrossRef.
- R. W. Read and R. M. Black, Rapid screening procedures for the hydrolysis products of chemical warfare agents using positive and negative ion liquid chromatography-mass spectrometry with atmospheric pressure chemical ionisation, J. Chromatogr., A, 1999, 862(2), 169–177 CrossRef CAS.
- D. D. Richardson and J. A. Caruso, Derivatization of organophosphorus nerve agent degradation products for gas chromatography with ICPMS and TOF-MS detection, Anal. Bioanal. Chem., 2007, 388(4), 809–823 CrossRef CAS.
- J. A. Caruso and M. Montes-Bayon, Elemental speciation studies—new directions for trace metal analysis, Ecotoxicol. Environ. Saf., 2003, 56(1), 148–163 CrossRef CAS.
- J. A. Caruso, et al., Modeling and separation–detection methods to evaluate the speciation of metals for toxicity assessment, J. Toxicol. Environ. Health, Part B, 2006, 9(1), 41–61 CAS.
- A. Montaser, Inductively Coupled Plasma Mass Spectrometry, Wiley-VCH, New York, 1998 Search PubMed.
- S. Wilbur, E. Soffy and E. McCurdy, Analysis of High Matrix Environmental Samples with the Agilent 7500ce ICP-MS with Enhanced ORS Technology, Agilent Application Note, Agilent Technologies, Inc., 2001, p. 5988-4286EN Search PubMed.
- J. Szpunar and R. Lobinski, Multidimensional approaches in biochemical speciation analysis, Anal. Bioanal. Chem., 2002, 373(6), 404–411 CrossRef CAS.
- K. M. Kubachka, et al., Detection of chemical warfare agent degradation products in foods using liquid chromatography coupled to inductively coupled plasma mass spectrometry and electrospray ionization mass spectrometry, J. Chromatogr., A, 2008, 1202(2), 124–131 CrossRef CAS.
- E. W. J. Hooijschuur, C. E. Kientz and U. A. T. Brinkman, Analytical separation techniques for the determination of chemical warfare agents, J. Chromatogr., A, 2002, 982(2), 177–200 CrossRef CAS.
- Y. R. Shu, et al., Screening of nerve agent degradation products by MALDI-TOFMS, Anal. Chem., 2006, 78(13), 4697–4701 CrossRef CAS.
- R. M. Black and R. W. Read, Application of liquid chromatography-atmospheric pressure chemical ionisation mass spectrometry, and tandem mass spectrometry, to the analysis and identification of degradation products of chemical warfare agents, J. Chromatogr., A, 1997, 759(1–2), 79–92 CrossRef CAS.
- D. R. Lide, Handbook of Chemistry and Physics, CRC Press, Cleveland, 78th edn, 1997–1998 Search PubMed.
- C. Pohl, Recent Developments in LC Column Technology, LC-GC, Europe, 2003, vol. 16(6a) Search PubMed.
- D. D. Richardson, B. B. M. Sadi and J. A. Caruso, Reversed phase ion-pairing HPLC-ICP-MS for analysis of organophosphorus chemical warfare agent degradation products, J. Anal. At. Spectrom., 2006, 21(4), 396–403 RSC.
| This journal is © The Royal Society of Chemistry 2010 |