Development and validation of an LC-MS/MS method for determination of phencyclidine in human serum and its application to human drug abuse cases
Krishna C.
Chimalakonda
a,
Chris
Hailey
b,
Ryan
Black
b,
Allison
Beekman
b,
Rebecca
Carlisle
b,
Elizabeth
Lowman-Smith
b,
Heather
Singletary
b,
S. Michael
Owens
c and
Howard
Hendrickson
*a
aDepartment of Pharmaceutical Sciences, College of Pharmacy, University of Arkansas for Medical Sciences, 4301 West Markham Street, Little Rock, AR 72205, USA. E-mail: hendricksonhowardp@uams.edu
bArkansas State Crime Laboratory, 3 Natural Resources Drive, P.O. Box 8500, Little Rock, AR 72215, USA
cDepartment of Pharmacology & Toxicology, College of Medicine, University of Arkansas for Medical Sciences, 4301 West Markham Street, Little Rock, AR 72205, USA
First published on 5th July 2010
Abstract
A new analytical method was developed and validated for the rapid determination of phencyclidine (PCP) in human blood and serum. Rapid chromatographic separation decreased the analysis time relative to standard gas chromatography (GC)-based methodologies. The method involved the use of solid-phase extraction for sample preparation and cleanup followed by liquid chromatography tandem spectrometric (LC-MS/MS) analysis and an electrospray-ionization (ESI) interface. PCP was quantified using multiple-reaction-monitoring with deuterium labeled PCP (PCP-d5) as an internal standard. The method was validated for accuracy, precision, linearity, and recovery. The method was accurate with error <14% and precision with coefficient of variation (CV) <5.0%. The assay was linear over the entire range of calibration standards (r2 > 0.997). The recovery of PCP after solid-phase extraction was greater than 90% with the lower limit of detection (LLOD) for PCP in 500 µl of human serum after solid-phase extraction at 0.06 ng ml−1. This method was used to determine the levels of PCP in postmortem human blood samples. The LLOD in blood was 1 ng ml−1. Blood PCP concentrations were also determined separately using GC and flame ionization detection (FID). Blood calibration standards and serum calibration standards yielded similar concentrations when used to quantitate authentic human blood samples that tested positive for PCP under the GC-FID method. Extraction of PCP from serum required fewer steps and therefore could be used as a calibration matrix in place of blood. The LC-MS/MS methodology shown here was higher throughput compared with GC-based methods because of very short chromatographic run times. This was accomplished without sacrificing analytical sensitivity.
A. Introduction
Phencyclidine, commonly called PCP, was synthesized in the early 1950s to be used as a surgical anesthetic.1 However, its therapeutic use was associated with the development of various side effects like blurred vision, delusions and problems associated with the normal activity of mental function, which led to its withdrawal from use as an anesthetic.2 Abuse of PCP began in the 1960s, and has been known by different street names like angel dust and peace pill. Prevalence of its abuse has never reached the level of marijuana, methamphetamine, or cocaine, but it remains a drug of abuse in the United States, with numerous overdose cases and occasional deaths.3 Though the abuse of PCP is limited relative to other illegal substances, toxicity is observed after very low doses of the drug. Low to moderate oral doses (5–20 mg) of PCP produces an acute, confused state that may last 6 hours. Higher inhaled doses (i.e., 2–20 mg) may cause adverse neurological or cardiovascular events and the person may be comatose for several days.4 A PCP cigarette may contain 5–50 mg of PCP. Though serum concentrations vary widely in patients experiencing PCP psychosis, levels of 5 ng ml−1 and below have been reported in some cases.5,6 Because PCP abuse is often associated with violent and destructive behavior, the impact of PCP on society as a whole is much greater than that observed with other drugs with higher abuse rates. Because PCP abuse does have a dramatic affect on human health, there are efforts to develop medications for the treatment of PCP abuse.7,8 The effects of therapeutic agents on the pharmacokinetics of PCP are critical towards the assessment of their safety and efficacy.The analytical methods of choice for the determination of PCP in biological fluids like blood, serum, plasma and urine are gas-chromatography-mass spectrometry9–14 or radio-immunoassay.6,15–19 Even though these methods are primary choices for PCP analysis, each one suffers from significant drawbacks. For radio-immunoassay, these include production of false-positive results due to problems of cross-reactivity and false-negative results due to inadequate sensitivity of the assay.20,21 Gas-chromatography-mass spectrometry (GC-MS) also suffers from certain limitations when used to assess clinical toxicology in patients suspected of PCP use. While it is true that most PCP related assays are developed to determine PCP concentrations postmortem, where cutoff values are typically set at 50 ng ml−1, there are situations where clinical toxicology is more important than the cause of death. In these clinical toxicology cases, concentrations less than 1 ng ml−1 are commonly found in patients and serum or plasma is normally the matrix used to determine PCP concentrations. Knowledge of whether a patient has been exposed to PCP will be more effective in treatment of the patient. While GC-MS has proven to effectively meet the needs of forensics and toxicology, it is still lacking with respect to the sensitivity and throughput required for clinical pharmacokinetic studies. The rate limiting step for GC-MS methods is a rather long chromatographic run time. Modern capillary GC columns produce PCP chromatograms with high separation efficiency, but require long temperature gradients to separate PCP from other components in the sample that interfere with the ionization of PCP. There is a lack of published methodologies for the determination of PCP in clinical pharmacokinetic samples. Though methods have been described for the determination of PCP pharmacokinetics in rats, these methods have not been validated in human samples.
Perhaps the most widely used biological fluid to detect PCP and other drugs of abuse has been plasma, serum and blood6,17,21–26 or urine.11,14,18,23,27,28 In the last 15 years, liquid chromatography with tandem mass spectrometric detection (LC-MS/MS) has been used extensively over methods like radio-immunoassay in the quantitation of various drugs of abuse like PCP in a variety of biological matrices. The advantages are improved sensitivity, greater specificity, rapid analysis and, most importantly, LC-MS/MS analysis reduces the possibility of inaccurate interpretation of the results due to false-negatives or false-positive findings.20,21 The lower cost and availability of LC-MS/MS equipment have made it possible for many clinical laboratories to have this instrumentation readily available. PCP and its metabolites have been quantitated in biological samples but there is a lack of a validated LC-MS method for the determination of PCP in whole blood obtained from human subjects.25–29 LC-MS/MS was used in a recent report to determine PCP in human urine.28 These investigators reported a lower limit of detection for PCP of 0.07 ng ml−1 when extracted from 500 µl of urine. In this report, PCP was determined along with 14 other known drugs of abuse. Because 14 other compounds were determined in a single chromatographic run, the total run time was 20 min. But these investigators did not show whether the run time could be shortened for PCP determination alone. In another recent report, Kala et al. have determined PCP in human oral fluid. Chromatographic run times were 6 min, but the LOD was only 2 ng ml−1.30
The primary aim of this work was to take full advantage of the selectivity and sensitivity of liquid chromatography with tandem mass spectrometry detection. When used in combination with a stable isotope internal standard we demonstrate a significantly decreased chromatographic run time relative to previously described methods for determination of PCP in biological fluids.
B. Experimental
1-[1-(Phenyl)cyclohexyl]piperidine hydrochloride (PCP-HCL) was obtained from the National Institute on Drug Abuse (Rockville, MD). The deuterated internal standard 1-[1-(phenyl-d5) cyclohexyl] piperidine (PCP-d5) was purchased from Sigma/Isotec (Miamisburg, OH). The chemical structure of phencyclidine (PCP) is shown in Fig. 1. Drug free pooled human serum was obtained from Chemicon/Millipore (Temecula, CA). Drug free human serum from individual donors was purchased from Innovative Research (Novi, MI). Drug free human whole blood was obtained from the Clinical Laboratory at the University of Arkansas Hospital (Little Rock, AR). Water for preparing mobile phase and other reagents was purified with a Milli-Q Synthesis A10 system (Millipore, Bedford, MA). Mobile phases were filtered using 0.2 µm nylon filters (Millipore) prior to use. LC-MS grade acetonitrile was purchased from Fisher Scientific (Houston, TX). All other chemicals were of analytical grade. | ||
| Fig. 1 Structure of phencyclidine (PCP). | ||
LC-MS/MS
A Quattro Premier triple-quadrupole mass spectrometer (Waters, Milford, MA) fitted with a Z-spray ion interface was used for all analyses. The LC system was a Shimadzu LC-10AD (Columbia, MD) equipped with a Shimadzu SIL-HT A autosampler. The LC system was interfaced to the mass spectrometer by means of an ESI source operated in the positive ionization mode.The liquid chromatographic separation of PCP and the internal standard PCP-d5 in the biological matrix was achieved on a Varian (Lake Forest, CA) 3 µm Pursuit C8 (100 × 2.0 mm, id) reversed-phase column preceded by a Varian MetaGuard 2.0 mm Pursuit 3 µm C8 guard column. Mobile phase A consisted of 20 mM ammonium formate (pH 2.70)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetonitrile (72:28%) and mobile phase B consisted of 20 mM ammonium formate (pH 2.70):acetonitrile (5:95%). Initial gradient conditions were held for 1 min at 10% B. The gradient was increased to 90% B (1–4 min) and then returned to initial conditions (4–8 min). The flow rate was 0.3 ml min−1 throughout the 10 min chromatographic run. The column temperature was maintained at 40 °C.
:acetonitrile (72:28%) and mobile phase B consisted of 20 mM ammonium formate (pH 2.70):acetonitrile (5:95%). Initial gradient conditions were held for 1 min at 10% B. The gradient was increased to 90% B (1–4 min) and then returned to initial conditions (4–8 min). The flow rate was 0.3 ml min−1 throughout the 10 min chromatographic run. The column temperature was maintained at 40 °C.
The following parameters were optimized for PCP MS analysis: capillary voltage, 3.0 kV; source block temperature, 120 °C; and desolvation gas (nitrogen) heated to a temperature of 450 °C and delivered at a flow rate of 550 l h−1. Multiple-reaction-monitoring (MS/MS) conditions were established at a cone voltage of 20 V. Collision-induced disassociation of each precursor ion was facilitated using argon at a pressure of 3.0 × 10−3 mbar and the collision energy was optimized to a value of 15 eV. The two ion transitions that were monitored for quantitation were m/z 244.2 → 86.2 for PCP and 249.3 → 86.3 for PCP-d5 with the interchannel delay and the interscan delay set to 0.05. The confirmation ion transition for PCP was m/z 244.2 → 158.8.
Sample preparation
Calibration standards (blank, 0.5, 1, 2, 5, 10, 20, 50, 100, 300, 500, and 1000 ng ml−1 PCP, calculated as a free base) were prepared in drug free human serum, or mobile phase A. Quality control standards (1, 20, and 800 ng ml−1) were prepared in serum, whole blood, and mobile phase A. Inter-day assay validation was performed using the same calibration standards and quality control samples on three different occasions.Sample preparation and cleanup was achieved using a Strata X-C Cation mixed-mode polymer (60 mg per 3 ml, Phenomenex, Torrance, CA) solid phase extraction (SPE) column. Serum (500 µl) was treated with 1.0 ml of 12% phosphoric acid and 50 µl of 10 ng ml−1 PCP-d5 and vortex-mixed for 5 s. The solid phase extraction (SPE) column was conditioned with 1.0 ml of methanol followed by 1.0 ml of 2% formic acid in water under gravity. The sample was loaded onto the SPE column and the flow rate was adjusted to 1–2 ml min−1 by vacuum or gravity. A 20-position vacuum manifold (Waters) was used to apply vacuum and to collect the eluent from the SPE cartridges. The SPE column was washed with 1.0 ml of 2% formic acid in water followed by 1.0 ml of methanol. The column was dried under high vacuum for 30 s and the drug was eluted with 5.0 ml of 10% v/v ammonium hydroxide in methanol. The eluent was dried under nitrogen at 40 °C and then reconstituted with 2.0 ml mobile phase A by: (1) vortex-mixing for 1.0 min, (2) allowed to standing at room temperature for 10 min, and (3) an additional vortex-mixing for 1.0 min. Attempts to reconstitute the sample in a smaller volume following SPE were also made. The dried residue was also dissolved in 50, 100, or 500 µl of mobile phase A. The sample (10.0 µl) was injected onto the analytical column for LC-MS/MS analysis. The sample loop volume was 100 µl.
Whole blood (500 µl) was first treated with 10% zinc sulfate (500 µl), centrifuged at 3000 rpm and the supernatant loaded onto a conditioned SPE cartridge. Further extraction of PCP from whole blood then proceeded as described above for serum. Fifteen samples could be managed with the vacuum manifold at one time.
Recovery and evaluation of matrix effects
PCP process recovery from serum and whole blood was determined from PCP spiked serum or whole blood (1, 300, and 1000 ng ml−1) which was then extracted and analyzed by LC-MS/MS. LC-MS/MS peak areas from these extracted samples were obtained. The process recovery was determined from ratio of the peak area for the standard prepared before extraction to that of the same standard prepared in mobile phase A. Process recovery was used here instead of extraction recovery because changes in the MS/MS response could be a result of sample loss from solid phase extraction or from matrix ion suppression.31Matrix ion suppression was evaluated using two approaches. Since a commercial source of human serum obtain from five individual donors was readily available, we employed a method described by Matuszewski.32 Matrix effects were deemed insignificant if calibration curves generated from each of these were not statistically different. Drug free serum from five different human donors was used to prepare standards for the generation of calibration slopes which were obtained by plotting the PCP concentration versus peak area. Whole blood from multiple donors was not readily available commercially, but pooled whole blood was easily obtained from the UAMS Clinical Laboratory (Little Rock, AR). Therefore a different approach was used to assess matrix ion suppression in whole blood. This second approach was also described by Matuszewski et al. and is summarized briefly below.33 Three sets of PCP standards were prepared. Set A consisted of PCP standards (1, 300, and 1000 ng ml−1) prepared neat in mobile phase A. Set B consisted of PCP standards (1, 300, and 1000 ng ml−1) prepared in whole blood before extraction. Set C consisted of PCP standards prepared in blood after extraction. Briefly, whole blood was extracted as described above and then spiked with PCP (1, 300, and 1000 ng ml−1) following the extraction. The ratio of spiked PCP peak area after extraction (Set C) to the peak area obtained from the same concentration in mobile phase A (Set A) was used to determine the matrix effects (MEs) in whole blood. Matrix effects in blood were expressed as this percentage. The extraction recovery was defined as the ratio of the peak area of the standards spiked before extraction to the peak area of the standards spiked after extraction.
Validation of the assay
Intra-day and inter-day validation was performed to evaluate the accuracy, precision, lower and higher limits of quantitation, and linear dynamic range. Assay validation in serum was performed by preparing a standard curve generated from 12 calibration standards (blank, 1, 2, 5, 10, 20, 50, 100, 300, 500, and 1000 ng ml−1 PCP, calculated as a free base) in drug free human serum and five sets of quality control samples at three different concentrations of 1, 20, and 800 ng ml−1. Calibration curves were generated by fitting linear regression analysis (with 1/x weighting) to the plot of PCP concentration versus the peak area ratio of PCP to PCP-d5. Assay validation in whole blood was performed by preparing a standard curve generated from four calibration standards (blank, 1, 300, and 1000 ng ml−1 PCP, calculated as a free base) in drug free human blood. Intra-day validation was performed by extracting the same set of calibration standards five times in a single day. The precision and accuracy were derived from coefficient of variation of predicted values and the ratio of the predicted value to the standard concentration × 100%. Inter-day assay validation was performed using the same calibration standards on three different occasions. The lower limit of detection was defined as the concentration that produced a signal-to-noise ratio (S/N) of 3. This was a concentration that was extrapolated from the S/N obtained at 1 ng ml−1. The S/N was determined by averaging the signal of extracted PCP (1 ng ml−1) standards (n = 5) and averaging the peak-to-peak noise observed in extracted PCP-free serum (n = 5).Analysis of authentic samples by LC-MS/MS
Postmortem blood from five different human subjects suspected of PCP intoxication was obtained from the Arkansas State Crime Laboratory (Little Rock, AR) and was analyzed for the presence of PCP using two different calibration matrices. Authentic whole blood samples, which were shown to contain PCP by a separate laboratory, were analyzed for PCP using either serum or whole blood as the calibration matrix. Samples were treated as described above in the validation section, except that serum standards were treated with the zinc sulfate solution. Calibration standards were also prepared in whole blood. Calibration standards were prepared in serum and blood and used to analyze authentic blood samples for PCP.Analysis of human blood by GC-FID
GC-FID analysis: gas chromatography with flame ionization detection (GC-FID) is an analytical method used routinely for confirmation of PCP in blood. It was used here to confirm the reliability of the LC-MS/MS method described above. The method described by Reynolds was used here, after minor modifications.34 Control samples were prepared by aliquoting 50 µl of PCP standard (0.1 g ml−1) and 50 µl of methaqualone (0.1 g ml−1) into 5 ml of PCP-free plasma. All retention times were locked to methaqualone to allow for long-term drift in the retention time. Before analysis by GC-FID, blood and urine samples (5 ml) were subjected to liquid–liquid extraction. The samples were first extracted with n-butyl chloride (10 ml). The n-butyl chloride was removed and treated with 5 ml of 1 M hydrochloric acid. The samples were mixed by rotation for 15 min and centrifuged for 5 min. The organic layer was discarded and the HCl layer treated with 1 ml of concentrated ammonium hydroxide. The two layers were thoroughly mixed and the HCl layer was then back-extracted into 100 µl of chloroform. The chloroform layer was transferred to an autosampler vial and 1 µl was injected onto the column for GC-FID analysis. Each authentic sample was analyzed one time to confirm the presence of PCP. Multiple assays of the same sample are not typically run by the Arkansas Crime Laboratory for postmortem cases so GC-FID data was not run more than once.GC-FID instrumentation
The GC system consisted of an Agilent 6890 gas chromatograph (Agilent Technologies, Santa Clara, CA) and a flame ionization detector. The analytical column was a 15 m × 0.32 mm Zebron ZB-5 column (Phenomenex, Torrance, CA). The film thickness was 0.25 µm. The split/splitless injector and detector were heated to 250 °C. Helium was used as the carrier gas with an inlet pressure of 7.5 psi. Splitless injection was used with purge time of 0.5 min. The solvent delay was set to 2.45 min. The initial temperature of the column was 100 °C and then programmed to achieve 280 °C at a rate of 15 °C min−1. The total run time was 18.5 min.C. Results and discussion
LC-MS/MS chromatograms of free extracted whole blood from a postmortem case suspected of PCP use and a calibration standard of PCP in extracted blood (1.0 ng ml−1) are shown in Fig. 2A and B respectively. These chromatograms were generated in MRM mode at the transitions of m/z 244.2 → 86.2 for PCP. The retention time of PCP was 1.90 min. Solid phase extraction recovery of PCP from serum and whole blood is shown in Table 1. The recovery from blood was approximately half that achievable in serum (51% versus 100%). To determine if this loss in PCP signal was due to poor recovery from the SPE cartridge or from matrix ion suppression of the MS/MS signal, extracted blood was spiked with PCP and these peak areas were compared to that obtained from the same concentration prepared in blood prior to SPE extraction. Data summarized in Table 2 show that the signal loss in PCP when extracted from blood was due to poor extraction recovery and not from matrix ion suppression. The higher than expected matrix effect observed at 1 ng ml−1 can be explained by the significant level of noise observed in drug-free extracted blood. The response was not adversely affected since the internal standard experienced the same noise.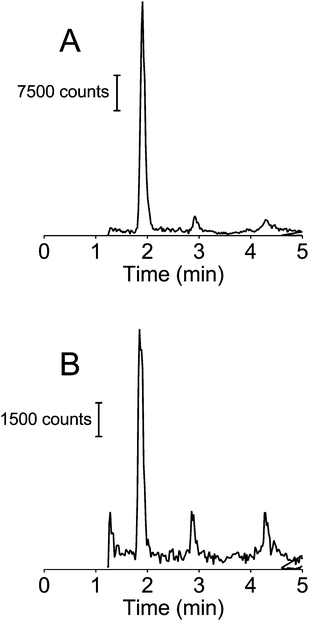 | ||
| Fig. 2 LC-MS/MS chromatograms of extracted whole blood from a suspected PCP user (A) and extracted serum spiked with 1 ng ml−1 PCP (B). | ||
| Nominal conc./ng ml−1 | Serum recovery (%) | Blood recovery (%) |
|---|---|---|
| 1 | 108 ± 11 | 54 ± 4 |
| 300 | 91 ± 10 | 39 ± 8 |
| 1000 | 102 ± 10 | 59 ± 3 |
| Average | 100 ± 9 | 51 ± 10 |
| Conc./ng ml−1 | Mean peak area (±CV)d | ME (C/A) (%) | RE (B/C) (%) | PE (B/A) (%) | ||
|---|---|---|---|---|---|---|
| Set Aa | Set Bb | Set Cc | ||||
| a Set A: PCP standards prepared in mobile phase A. b Set B: PCP standards prepared in whole blood before extraction. c Set C: PCP standards prepared in whole blood after extraction. d Values are the Mean ± Coefficient of variation (n = 5). | ||||||
| 1 | 2413 (±1) | 1384 (±12) | 3639 (±2) | 150 | 38 | 57 |
| 300 | 743378 (±1) |
284428 (±4) |
901413 (±2) |
121 | 32 | 38 |
| 1000 | 2359031 (±1) |
995036 (±1) |
2422714 (±2) |
103 | 41 | 42 |
Matrix effects originating from extracted serum did not adversely affect the recovery of PCP. This is clearly shown by the excellent process recovery (100 ± 9%) of PCP from serum and the reproducibility of the calibration curves generated in serum from five different donors (CV = 9%)
Excellent accuracy of the assay was demonstrated by error values of ≤14.0% for all the concentrations, including the lowest calibration curve concentration (1 ng ml−1). The precision was shown by CV values <11.0% for the three quality control serum standards. The intra-day precision for PCP in blood was 7%, 11%, and 5% at 1, 20, and 1000 ng ml−1.
Excellent inter-day accuracy of the assay was demonstrated by error values of <5.0% for all the serum standard concentrations. The assay was also deemed precise, as shown by CV values <5.0% for 20 and 800 ng ml−1 and <16.0% for the lowest concentration of 1.0 ng ml−1. The accuracy was within 80–120% of the nominal value and the CV <20% at 1 ng ml−1, the lowest calibration standard. This excellent inter-day accuracy and precision at 1 ng ml−1, therefore, predict a lower limit of quantitation of 1 ng ml−1. Additionally, the relationship between peak area ratios of PCP and the detector response was linear (r2 ≥ 0.997) over the studied concentration range of 1–1000 ng ml−1 for both intra-day and inter-day validation. The lower limit of detection in serum was 0.06 ng ml−1 (500 µl of serum) or 0.6 pg on-column. Attempts to improve the LLOD by decreasing the final volume following SPE did not result in an increase in the LC-MS/MS signal. In fact there was a significant decrease in the signal when compared to the peak area obtained when 2 ml was used to reconstitute the sample (data not shown). Reasons for this observation may be either incomplete dissolution of the analyte into the smaller volume or an increase in the concentration of ion suppressing components in the serum.
Determination of PCP concentrations in postmortem blood obtained from cases of suspected PCP intoxication further tested the robustness and utility of this assay. Significant levels of PCP were quantitated in blood, with the concentration of PCP ranging from 3.2–80.0 ng ml−1 in blood (Table 3). The lower limit of detection in blood was 1 ng ml−1 (S/N = 3) or 10 pg on-column. Although the extraction recovery for PCP and PCP-d5 was significantly lower in whole blood than in serum, serum and blood worked equally well as a calibration matrix for quantitation of PCP in whole blood. Since serum and blood functioned equally well as calibration matrix, the method has been effectively validated for blood and serum. Furthermore, these results show that serum can serve as a matrix for PCP determinations in blood. Serum was a much easier matrix to prepare for LC-MS/MS analysis and can therefore improve the throughput when the authentic samples are only available in blood, since calibration standards can be prepared in serum.
Comparison of the LC-MS/MS method to the GC-FID method showed reasonable agreement between the two methods. The slope of LC-MS/MS values versus those obtained by GC-FID was 1.4 (r2 = 0.87) when serum was used as the calibration matrix and 1.3 (r2 = 0.89) when blood was the calibration matrix for LC-MS/MS.
D. Conclusions
The main objective of the study was to develop an analytical method to quantitate phencyclidine (PCP) in human serum and whole blood in order to demonstrate the speed and sensitivity of LC-MS/MS. We have also further validated the method by using a second analytical method (i.e., GC-FID) to confirm PCP concentrations in authentic blood samples. While the sample preparation time was similar to that reported for GC-MS, the chromatographic run time and lower limit of quantitation demonstrated that LC-MS/MS is more applicable to pharmacokinetic studies in humans than the well established GC-MS methods. In part, the reason GC-MS has not been surpassed by LC-MS/MS for the routine analysis of PCP in human subjects is most of the interest in human PCP use has been to evaluate patients for PCP intoxication. There have been several recent reports of PCP determinations in biological fluids using LC-MS/MS. Feng et al. have reported an LOD of 0.07 ng ml−1 for PCP extracted from 500 µl of urine, which was very similar to the LOD of 0.06 ng ml−1 reported herein.28 But the chromatographic run time in the methodology described by Feng et al. was 20 min, which was significantly longer than the 10 min run time described here. Conversely, Kala et al. have described LC-MS/MS methodology for PCP determinations in oral fluid with a 6 min chromatographic run time.30 But Kala and co-workers could only achieve an LOD of 2 ng ml−1 from 1 ml of oral fluid. We have also further validated the method by using a second analytical method (i.e., GC-FID) to confirm PCP concentrations in authentic blood samples.There has been significant progress made in the development of medications for the treatment of PCP abuse. New analytical methods are needed to evaluate the safety and efficacy of the medication and their effects on the pharmacokinetics of PCP. The LC-MS/MS method described herein may be useful in the determination of PCP pharmacokinetic samples where the PCP dose is significantly lower than that typically met with following PCP intoxication or overdose.
Acknowledgements
Funding for this work was from NIDA/NIH (R42DA017596 and R01DA007610) and the University of Arkansas for Medical Sciences College of Pharmacy.References
- A. J. Catenacci, D. D. Grove, W. A. Weiss, S. M. Fisher, A. M. Sismondo and J. H. Moyer, Antibiot. Med. Clin. Ther. (N. Y.), 1959, 6, 145–150 Search PubMed.
- E. F. Domino, Int. Rev. Neurobiol., 1964, 6, 303–347 CAS.
- A. Mozayani, Forensic Sci. Rev., 2003, 15, 61–74 Search PubMed.
- G. D. Lundberg, R. C. Gupta and S. H. Montgomery, Clin. Toxicol., 1976, 9, 503–511 Search PubMed.
- D. C. Javitt and S. R. Zukin, Am. J. Psychiatry, 1991, 148, 1301–1308 Search PubMed.
- C. B. Walberg, M. M. McCarron and B. N. Schulze, J. Anal. Toxicol., 1983, 7, 106–110 CAS.
- H. M. Lacy, M. G. Gunnell, E. M. Laurenzana and S. M. Owens, Int. Immunopharmacol., 2008, 8, 1–11 CrossRef CAS.
- S. Castellani, P. M. Adams and A. J. Giannini, J. Clin. Psychiatry, 1982, 43, 10–11 Search PubMed.
- S. Schneider, P. Kuffer and R. Wennig, J. Chromatogr., B: Biomed. Sci. Appl., 1998, 713, 189–200 CrossRef CAS.
- D. Cox, R. A. Jufer Phipps, B. Levine, A. Jacobs and D. Fowler, J. Anal. Toxicol., 2007, 31, 537–539 CAS.
- P. M. Froehlich and G. Ross, J. Chromatogr., 1977, 137, 135–143 CrossRef CAS.
- C. G. Hammar, B. Holmstedt, J. E. Lindgren and R. Tham, Adv. Pharmacol. Chemother., 1969, 7, 53–89 Search PubMed.
- D. Legault, J. Chromatogr., 1980, 202, 309–312 CrossRef CAS.
- C. C. Stevenson, D. L. Cibull, G. E. Platoff, Jr, D. M. Bush and J. A. Gere, J. Anal. Toxicol., 1992, 16, 337–339 CAS.
- M. S. Swartz, J. W. Swanson and M. J. Hannon, Psychiatr. Serv., 2003, 54, 891–895 CrossRef.
- B. Kaul and B. Davidow, Clin. Toxicol., 1980, 16, 7–15 Search PubMed.
- S. M. Owens, J. Woodworth and M. Mayersohn, Clin. Chem., 1982, 28, 1509–1513 CAS.
- S. J. Mule and G. A. Casella, J. Anal. Toxicol., 1988, 12, 102–107 CAS.
- K. A. Moore, C. Werner, R. M. Zannelli, B. Levine and M. L. Smith, Forensic Sci. Int., 1999, 106, 93–102 CrossRef CAS.
- A. E. Davis and M. Peat, Drug Abuse Handbook, CRC Press, 1998, pp. 751–764 Search PubMed.
- A. G. Verstraete and F. V. Heyden, J. Anal. Toxicol., 2005, 29, 359–364 CAS.
- S. S. Tai, J. L. Prendergast, L. T. Sniegoski, M. J. Welch, K. W. Phinney and N. F. Zhang, Anal. Bioanal. Chem., 2010, 397, 501–509 CrossRef CAS.
- A. M. Ferguson and U. Garg, Methods Mol. Biol., 2010, 603, 461–467 CAS.
- J. R. Woodworth, M. Mayersohn and S. M. Owens, J. Anal. Toxicol., 1984, 8, 2–6 CAS.
- H. P. Hendrickson, E. C. Whaley and S. M. Owens, J. Mass Spectrom., 2005, 40, 19–24 CrossRef CAS.
- M. Sergi, E. Bafile, D. Compagnone, R. Curini, G. D'Ascenzo and F. S. Romolo, Anal. Bioanal. Chem., 2009, 393, 709–718 CrossRef CAS.
- S. Kerrigan and P. J. W. H. Jr, Clin. Chem., 2001, 47, 540–547 CAS.
- J. Feng, L. Wang, I. Dai, T. Harmon and J. T. Bernert, J. Anal. Toxicol., 2007, 31, 359–368 CAS.
- C. Coulter, K. Crompton and C. Moore, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2008, 863, 123–128 CrossRef CAS.
- S. V. Kala, S. E. Harris, T. D. Freijo and S. Gerlich, J. Anal. Toxicol., 2008, 32, 605–611 CAS.
- A. K. Majumdar, J. B. McCrea, D. L. Panebianco, M. Hesney, J. Dru, M. Constanzer, M. R. Goldberg, G. Murphy, K. M. Gottesdiener, C. R. Lines, K. J. Petty and R. A. Blum, Clin. Pharmacol. Ther., 2003, 74, 150–156 CrossRef CAS.
- B. K. Matuszewski, J. Chromatogr., B: Biomed. Appl., 2006, 830, 293–300 CrossRef CAS.
- B. K. Matuszewski, M. L. Constanzer and C. M. Chavez-Eng, Anal. Chem., 2003, 75, 3019–3030 CrossRef CAS.
- P. C. Reynolds, Clin. Toxicol., 1976, 9, 547–552 Search PubMed.
| This journal is © The Royal Society of Chemistry 2010 |