DOI:
10.1039/C0AY00215A
(Paper)
Anal. Methods, 2010,
2, 1144-1151
A simple and efficient approach to improve protein identification by the peptide mass fingerprinting method: concomitant use of negative ionization
Received
1st April 2010
, Accepted 12th May 2010
First published on
10th June 2010
Abstract
Peptide mass fingerprinting (PMF) has been widely used as an efficient analytical strategy for protein identification. This is most commonly used with a combination of protein digestion using sequence-specific proteases and MALDI-TOFMS. Then database searches are performed comparing the pattern of the experimentally obtained masses with the pattern of the theoretical peptide masses of proteins stored in the database. The positive ionization mode has been mainly used for MALDI analyses with a few exceptions for phosphopeptides, oligonucleotides, etc. Therefore, nonphosphorylated peptides that have low pI values could be missed from PMF using the positive ionization mode. Here, we introduce optimal conditions for negative ionization of peptides and the practical advantages of negative ionizations in PMF. Angiotensin I (pI 6.9) and bovine serum albumin (BSA) tryptic digests were used as model peptides. Eight matrix candidates and seven additives were examined in terms of sensitivity, robustness and reproducibility. The combination of DHB and phosphoric acid was the best condition for negative ionization of peptides and was found to be compatible with the positive ionization mode. Using 150 mM DHB/1% phosphoric acid, the coverage (% by amino acid count) of BSA tryptic digest (0.6 pmol per spot) totaled 67.2% (negative + positive). The 24.1% of peptides (pI range 4.1–6.2) were detected only by negative ionization, which indicated that acidic peptides were efficiently recovered by the negative ion mode. This methodology has been successfully employed to analyze other proteins without false positive identifications.
Introduction
Mass spectrometry (MS) has become a powerful tool in the field of biochemistry and biology since the soft ionization methods of electrospray ionization and laser desorption ionization were developed by Fenn1 and Tanaka,2 respectively. In particular, protein identification now exclusively relies on the use of MS coupled with the above two ionization techniques. The strategy called “proteomics” therefore has spread with the high expectation3 that it could express phenotypes under various physiological states as the next era of genomics.4 Peptide mass fingerprinting (PMF) is one of the main methods in proteomics and has been widely used as an efficient analytical strategy for protein identification.5 This is most commonly used with a combination of protein digestion using sequence-specific proteases and matrix-assisted laser desorption ionization (MALDI)-time of flight (TOF)MS. Database searches are performed comparing the pattern of the experimentally obtained masses with the pattern of the theoretical peptide masses of proteins stored in the database.
For MALDI-TOFMS analyses, positive ionization to produce protonated molecules has been mainly used. For instance, α-cyano-4-hydroxycinnamic acid (CHCA) and 2,5-dihydroxybenzoic acid (DHB) have been used exclusively as matrices for PMF to produce protonated peptides. On the other hand, there are several studies that have reported the use of the negative ionization mode for compounds that produce negatively charged functional groups. Fitzgerald et al. have screened substituted pyrimidine, pyridine and benzene derivatives containing basic amino groups as potential matrices for negative ionization of proteins and oligonucleotides.6 Also, Vermillion-Salsbury and Hercules have used 9-aminoacridine (9-AA) as a matrix for negative ionization of small molecules and oligonucleotides.7 Shroff et al. also have shown the usefulness of 9-AA for low molecular weight acids.8 Nonami et al. have reported β-carboline alkaloids for oligosaccharides,9 and pyridoindoles, pyridylindoles and pyridylpyridoindoles for proteins,10 as the basic matrices.
The existence of alkali metal ions normally creates critical problems in both negative and positive ionization modes of MALDI, such as the formation of adduct ions, suppression of ionization, etc. Therefore, several attempts to eliminate the alkali metal ions to improve MALDI analyses have been reported. Smirnov et al. have reported that washing the sample/matrix spot using aqueous ammonium phosphate improved the sensitivity.11 The usefulness of matrix additives has also been reported as another approach. For oligonucleotides, tetraamines such as spermine12 and alkali metal ionophores such as crown ethers13 have been reported. In the proteomics field, the issue of phosphorylated peptide and protein analyses has been well studied because of their biological significance. However, there are more complicated analytical difficulties, mainly resulting from the low abundance of phosphorylation and the formation of ion pairs with alkali metal ions. Diammonium citrate with CHCA or DHB,14,15 phosphoric acid with DHB or CHCA,16 alkylphosphonic acid with DHB,17 and monoammonium phosphate with CHCA18 have been reported to enhance sensitivities of phosphorylated peptides and proteins. Because phosphorylated peptides have been shown to favor negative ion production,19 some of the above additives were also effective in negative ionization mode.16,17
As mentioned, negative ionization aspects in proteomics are normally limited to the analyses of phosphorylated peptides after eliminating the effects of alkali metal ions. Therefore, only a few aspects have been shown for intact (nonphosphorylated) peptides. Nishikaze and Takayama have shown that adding an amino acid, serine, to the CHCA matrix was effective in removing interfering alkali metal from intact peptides, especially for the negative ion mode.20 Recently, DHB was also shown by Gao and Cassady to be a good negative matrix for neutral peptides and proteins.21 CHCA and DHB are typical proton donors and favor positive ion production, but it is interesting to observe negative ions from intact peptides. Therefore, the potential usefulness of negative ionization for proteomics should be considered not only for phosphorylated peptides, but also for low pI intact peptides. Nabetani et al. have shown the usefulness of negative ionization to identify sulfonated proteins as well as phosphorylated proteins from 2-dimentional electrophoresis samples.22 Bigwarfe and Wood have explored both MALDI and electrospray ionization, and have obtained better sequence coverage from model proteins by combining the positive and negative ionization data.23 Additionally from the viewpoint of instrumentation, Tsai et al. have reported dual polarity TOFMS, which enables simultaneous MS analyses.24 These aspects have suggested us to revisit the concomitant use of negative ionization PMF to build a practical and robust way to improve protein identification (Fig. 1). The database search is based on the pattern analyses without any sequence information; therefore, it is ideal to ionize whole digested peptides efficiently that have diverse pI values to improve the identification based on the similarity to the theoretical peptide masses of proteins (Fig. 1a). In addition, finding common proteins identified from both positive and negative ion modes would be useful to avoid false positive identifications from complicated samples (Fig. 2a). Here, we first optimized the conditions for negative ionization of intact peptides, and introduced the practical advantages of the negative ionization to improve protein identification by PMF.
 |
| | Fig. 1 Concept of the practical advantage of negative ionization in PMF. | |
 |
| | Fig. 2 Matrices and additives used in this study. Matrices: α-cyano-4-hydroxycinnamic acid (CHCA, a), 2,5-dihydroxybenzoic acid (DHB, b), nor-harman (NH, c), 9-aminoacridine (9-AA, d), 4-nitroaniline (NA, e), 3-methyl-4-nitroaniline (MNA, f), 2-amino-5-nitropyridine (ANP, g) and 2-amino-4-methyl-5-nitropyridine (AMNP, h). Additives: 4′-nitrobenzo-15-crown-5 ether (NBC, i), N,N′-dibenzyl-4,13-diaza-18-crown-6 ether (DDC, j), phosphoric acid (PA, k), methylenediphosphonic acid (MDPNA, l), L-serine (Ser, m), L-threonine (Thr, n) and L-arginine (Arg, o). | |
Experimental
Chemicals and materials
Ammonium bicarbonate, iodoacetamide (IAA), dithiothreitol (DTT), phosphoric acid (PA), methylenediphosphonic acid (MDPNA), L-serine (Ser), L-threonine (Thr), L-arginine (Arg) and urea were purchased from Nacalai Tesque, Inc. (Kyoto, Japan). 2,5-Dihydroxybenzoic acid (DHB), 9-aminoacridine (9-AA), 4-nitroaniline (NA), 3-methyl-4-nitroaniline (MNA), 2-amino-5-nitropyridine (ANP), 2-amino-4-methyl-5-nitropyridine (AMNP), 4′-nitrobenzo-15-crown-5 ether (NBC) and N,N′-dibenzyl-4,13-diaza-18-crown-6 ether (DDC) were purchased from Tokyo Chemical Industry Co. Ltd. (Tokyo, Japan). Trifluoroacetic acid (TFA) and formic acid were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). HPLC grade acetonitrile was obtained from Kanto Chemical Co. Inc. (Tokyo, Japan). Purified water was purchased from Daiwa-Yakuhin Co. Ltd. (Sendai, Japan) and further filtered through Ultrapure Water System, CPW-100 (Advantec Toyo Kaisha Ltd., Tokyo, Japan). α-Cyano-4-hydroxycinnamic acid (CHCA), nor-harman (NH), bovine serum albumin (BSA), human serum albumin (HSA), transferrin, lysozyme, α-casein and adrenocorticotropic hormone (ACTH) (18–39) (RPVKVYPNGAEDESAEAFPLEF) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Angiotensin (Ang) I (DRVYIHPFHL) was purchased from American Peptide Company, Inc. (Sunnyvale, CA, USA). Human Ang II (DRVYIHPF) was obtained from Calbiochem/EMD Chemicals Inc. (San Diego, CA, USA). Sequencing grade trypsin was purchased from Promega Co. (Madison, WI, USA). ZipTipC18 cartridges were obtained from Millipore Co. (Bedford, MA, USA).
MALDI-TOFMS
MALDI-TOFMS experiments were carried out using a Voyager-DE STR MALDI-TOF mass spectrometer (Applied Biosystems, Framingham, MA, USA) located in the Biomedical Research Core, School of Medicine, Tohoku University. The mass spectrometer was equipped with a nitrogen laser (337 nm, 3 ns pulse width, 20 Hz repetition rate) and has a flight path of 200 cm. All of the spectra presented were acquired in reflectron mode. The mass spectrometer parameters were optimized to obtain maximum signal to noise ratio (S/N). Typical parameters were as follows: accelerating voltage, 20 kV; grid voltage, 64%; extraction delay time, 100 ns; low mass gate, 500 Da; shots in spectrum, 100; mass range, m/z 500–4000. All acquired data were processed using Data Explorer Version 4.0.0.0.
Optimization for negative ionization using Ang I
Ang I (1.3 mg) was dissolved in water (100 µL) followed by serial dilution to yield a solution of 2 pmol µL−1. Aliquots (0.5 µL, 1 pmol) were spotted on a 96-well MALDI sample plate. Each matrix solution containing internal calibrants (1 pmol Ang II, [M + H]+ at m/z 1046.5423, [M − H]− at m/z 1044.5267; 1 pmol ACTH (18–39), [M + H]+ at m/z 2465.1989, [M − H]− at m/z 2463.1832) was prepared in 50% (w/w) aqueous acetonitrile and 0.5 µL of the solution was mixed with Ang I solution on a MALDI plate and allowed to dry at room temperature. Because the MALDI signal intensity is dependent on the irradiation position, the best spot was found manually, and the data were generated as an average of shots. The sensitivity was estimated from the S/N value, but robustness (intensity) and reproducibility were also considered, if they were critical for practical use.
MALDI-TOFMS analyses of BSA tryptic digestion
BSA (0.5 mg) was dissolved in 6.5 M urea (500 µL). An aliquot (20 µL) of the solution was placed in a centrifuge vial. DTT (110 mM) in 12.5 mM ammonium bicarbonate (2 µL) was added to the tube and the solution was incubated at 37 °C for 1 h to reduce the disulfide bonds. The reduced cysteines were then alkylated by the addition of 600 mM IAA in 12.5 mM ammonium bicarbonate (2 µL) and incubated at room temperature in the dark for 45 min. The reduced and alkylated BSA was diluted with 12.5 mM ammonium bicarbonate (72 µL) and an aliquot (40 µL, corresponding to 8.33 µg of BSA) digested using sequencing grade trypsin (0.1 µg mL−1, 4 µL, enzyme : protein = 8.33 : 0.4, w/w) and incubated overnight at 37 °C. The pH of the digested protein sample was then lowered to 3 with 50% (v/v) aqueous formic acid solution (4 µL). The solution was desalted using a ZipTipC18 cartridge. Briefly, the ZipTip was conditioned with 75% (v/v) aqueous acetonitrile (10 µL) and 0.1% (v/v) TFA in water (10 µL). The BSA tryptic digest was subsequently extracted from the solution (BSA 8.33 µg per 48 µL) by the ZipTip, and washed with 0.1% aqueous TFA (10 µL). Finally, the peptides were eluted with 75% aqueous acetonitrile containing 0.1% TFA (10 µL). The eluate was kept as a BSA tryptic digest standard solution (×1) and the concentration was estimated as 8.33 µg (124 pmol) per 10 µL (as 100% recovery from ZipTip). For MALDI-TOFMS analyses, the standard solution was used as it is or after dilution of ×10, ×20, ×100 and ×200. Aliquots (0.5 µL) were spotted on a 96-well MALDI sample plate followed by 0.5 µL of matrix solution of saturated CHCA in water : acetonitrile : TFA (50 : 50 : 0.1, v/v/v) or 150 mM DHB in water : acetonitrile : PA (50 : 50 : 1, v/v/v) containing internal calibrants (1 pmol Ang II, [M + H]+ at m/z 1046.5423, [M − H]− at m/z 1044.5267; 1 pmol Ang I, [M + H]+ at m/z 1296.6853, [M − H]− at m/z 1294.6697; and 1 pmol ACTH (18–39), [M + H]+ at m/z 2465.1989, [M − H]− at m/z 2463.1833) and allowed to dry at room temperature.
Applications for other proteins
HSA, transferrin, lysozyme, and α-casein (20 µg) were digested in the same manner written in the BSA section. The samples were diluted with water and analyzed by MALDI-TOFMS.
Database search criteria
Peaks used for PMF were monoisotopic peaks that had S/N > 20. Peptide coverage was carried out using a PMF tool, Aldente in the ExPASy (Expert Protein Analysis System) proteomics server of the Swiss Institute of Bioinformatics (SIB) (http://www.expasy.org/tools/aldente/) with the following criteria: database, UniProtKB/Swiss-Prot, Release 56.9 of 03-Mar-2009; Enzyme, trypsin; Missed cleavage, 1; Resolution, Monoisotopic; Modifications, carbamidomethyl (Cys), oxidation (Met), phosphorylation (Ser, Thr and Tyr); Spectrometer shift max, ±0.2 Da; Spectrometer slope max, ±200 ppm; Spectrometer internal error max, ±25 ppm; Minimum number of hits, 4. The sequence coverage was based on the number of amino acids.
Estimation of false positive identifications
Tryptic peptides from HSA (100 fmol), transferrin (50 fmol) or α-casein (100 fmol) standard were analyzed by MALDI-TOFMS and searched as unknown proteins using the criteria described in the previous section. Additional criteria were applied as follows: species, Homo sapiens; Mw range, 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–70000; pI range, 5–7 (for HSA); species, H. sapiens; Mw range, 60000–80000; pI range, 5–7 (for transferrin); species, Bos taurus; Mw range, 20000–40000; pI range, 4–7 (for α-casein).
000–70000; pI range, 5–7 (for HSA); species, H. sapiens; Mw range, 60000–80000; pI range, 5–7 (for transferrin); species, Bos taurus; Mw range, 20000–40000; pI range, 4–7 (for α-casein).
Results and discussion
Matrices for negative ionization
Two acidic matrices (CHCA and DHB) and six basic matrices (NH, 9-AA, NA, MNA, ANP and AMNP) were tested as suitable candidates for negative ionization (Fig. 2a–o). To optimize conditions, Ang I (DRVYIHPFHL) was used as a model peptide. Ang I was selected by consideration of its neutral pI (6.9), which could be useful to see the balance between negative and positive sensitivities. The optimal conditions were chosen in terms of the sensitivity (S/N), robustness (signal intensity) and reproducibility. The optimal concentration for each matrix was determined according to its solubility as follows: CHCA (20 mM saturated), DHB (50–200 mM), NH (10–100 mM), 9-AA (2–20 mM), NA (50–100 mM), MNA (50–100 mM), ANP (50–100 mM) and AMNP (50–100 mM). The S/N values for the optimal condition and crystal form of each matrix are summarized in Fig. 3. Among them, 150 mM DHB and 20 mM NH gave relatively good S/N (>200). In terms of robustness and reproducibility, the matrix concentration was critical for the signal of NH. Nitrobenzene derivatives (ANP and AMNP) were dependent on laser irradiation position, probably because of lower homology of the crystallization.
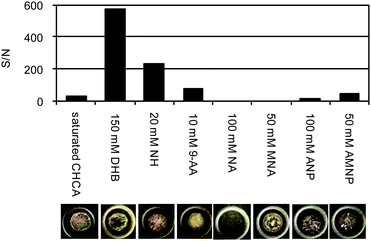 |
| | Fig. 3 Effect of matrices on S/N and the crystal forms. The optimal concentration of each matrix was examined in the preliminary experiment according to its solubility. Sample: Ang I, 1 pmol per spot. | |
Effect of additives
The effect of alkali metals has already been pointed out, especially on the analyses of oligonucleotides. Formation of Na/K adduct ions and suppression of ionization always reduce sensitivity in negative ionization mode. Therefore, several reagents were tested as additives for 150 mM DHB, saturated CHCA and 20 mM NH (Fig. 4). Adding PA gave a significant improvement in the sensitivities for DHB (Fig. 4a, ×3, S/N ≈ 1800) and CHCA (Fig. 4b, ×6, S/N ≈ 180); however, there was no effect for NH (Fig. 4c). The amino acids also increased S/N for all three of them, but their effects were not as significant as those of PA. Unexpectedly, crown ethers, known as alkali metal ionophores, were not effective additives for any of the three. Overall, 150 mM DHB with 1% PA gave the best result for Ang I. The effect of PA on DHB was reported for negative ionization of phosphorylated peptides and proteins by Kjellström and Jensen,16 but these data suggest that it also works for neutral peptides such as Ang I (pI 6.9).
 |
| | Fig. 4 Effect of alkali metal trapping reagents: (a) 150 mM DHB, (b) saturated (∼100 mM) CHCA, (c) 20 mM NH. Sample: Ang I, 1 pmol per spot. | |
Effect of PA on Ang I doped with 25 mM NaCl
Because adding PA was effective in improving negative ionization, 25 mM NaCl-doped Ang I sample was analyzed to see the effect (Fig. 5). Without PA, the ion resulted from sodium ([M + Na − 2H]− at m/z 1316.67) was observed as ∼20% intensity of the deprotonated ion ([M − H]− at m/z 1294.67) (Fig. 5a). The addition of 1% PA completely eliminated the formation of the sodium adduct with increases in the signal intensity (from 1.3 × 104 to 1.7 × 104) and the S/N value (from 499 to 730) (Fig. 5b). Interestingly, this indicates that the removal of alkali metals was more effective in ionizing peptides negatively rather than the basic atmosphere.
 |
| | Fig. 5 MALDI-TOFMS spectra of Ang I doped with 25 mM NaCl: (a) 150 mM DHB without PA, (b) 150 mM DHB with PA (1%, v/v). Sample: Ang I, 1 pmol per spot. | |
Comparison of the amino acid sequence coverage using negative and positive ion modes
For negative ionization of peptides, the combination of DHB and phosphoric acid was optimal in terms of sensitivity, robustness and reproducibility. Because this combination also gave good sensitivity in the positive ionization mode, it can be used as a compatible matrix for both negative and positive ionization data. This can also be a big advantage in obtaining negative and positive ionization data by a single sample preparation. Therefore, BSA tryptic digest (0.6 pmol per spot) was analyzed with both ionization modes (Fig. 6). The peptide coverage was compared between the data obtained by positive (Fig. 6a) and negative ionizations (Fig. 6b). Acidic peptides (pI values ≈ 4), which were not detected by positive ion mode, were efficiently recovered by negative ion mode, as we expected. Therefore, in the use of only positive ionization mode, not only phosphorylated peptides, but also intact peptides that have low pI, could be missed.
 |
| | Fig. 6 MALDI-TOFMS spectra of BSA tryptic digest. (a) Positive ion mode with 150 mM DHB/1% PA, (b) negative ion mode with 150 mM DHB/1% PA. Sample: BSA tryptic digest, 0.6 pmol per spot. Asterisk indicates the peak from an internal calibrant (Ang II). The pI value of each peptide is shown in tables. | |
Effect of concentration on the coverage
Because concomitant use of negative and positive ion modes with 150 mM DHB/1% PA was found to be a powerful way to increase the peptide coverage, the coverage (% by amino acid count) of BSA tryptic digest was determined at several concentrations (Fig. 7). At the highest concentration examined (×1, 6 pmol per spot), the coverage (% by amino acid count) was as follows: detected by positive ion only, 3.1%; detected by negative ion only, 17.0%; detected by both ionizations, 60.1%. At the lowest concentration used (×100, 60 fmol per spot), the coverage was as follows: detected by positive ion only, 6.2%; detected by negative ion only, 8.1%; detected by both ionizations, 4.3%. Therefore, the concomitant use of negative ionization was more efficient for lower concentration samples to increase the coverage; for the sample of ×1, from 63.2% (3.1% + 60.1%) to 80.2%; for the sample of ×100, from 10.5% (6.2% + 4.3%) to 18.6%. Compared with a typical PMF condition, saturated CHCA/0.1% TFA, 26.4% and 0% of peptides were found by positive and negative ionization modes, respectively. The pI range of peptides found with CHCA positive ionization mode was 4.4–8.8.
 |
| | Fig. 7 Recovery of tryptic peptides using different sample concentrations. Both positive and negative ion modes with 150 mM DHB/1% PA. Sample: BSA tryptic digest, 30 fmol (×200) to 6 pmol (×1) per spot. | |
Applications for other proteins
To confirm the strategy, several proteins were analyzed by the method we have developed (Table 1). The model proteins were chosen from the viewpoints of different pI, hydrophobicities, etc. Phosphrylated peptides were found from α-casein digest only with negative mode as we expected. Overall, acidic peptides and basic peptides tended to be detected by negative ionization and positive ionization, respectively. However, there are some exceptions found especially from longer peptides, such as, V265HTECCHGDLLECADDRADLAK286 (from HSA, pI 4.5, positive only), K122DSGFQMNQLR132 (from transferrin, pI 8.7, negative only), H554QTVPQNTGGKNPDPWAK571 (from transferrin, pI 8.6, negative only), V20FGRCELAAAMK31 (from lysozyme, pI 8.2, negative only), D58IGpSEpSTEDQAMEDIK73 (from α-casein, pI 3.7, positive only), etc. These phenomena could be resulted from the intramolecular ion pair formation.
Table 1 Tryptic peptides detected (S/N > 20) from HSA (10 fmol), transferrin (10 fmol), lysozyme (50 fmol), and α-casein (50 fmol). Asterisks indicate modifications, oxidation (M), phosphorylation (S, T and Y). The sequence coverage was based on the number of amino acids
| Proteins |
Peptides detected |
m/z positive/negative |
pI |
Detected |
Coverage (%) |
| Positive |
Negative |
Positive |
Negative |
Total |
| HSA (10 fmol) |
F35KDLGEENFK44 |
1226.6/1224.6 |
4.7 |
○ |
○ |
26.2 |
33.7 |
40.1 |
| L66VNEVTEFAK75 |
1149.6/1147.6 |
4.5 |
○ |
○ |
| S89LHTLFGDKLCTVATLR105 |
1932.0/1930.0 |
8.0 |
○ |
○ |
| Y162LYEIAR168 |
927.5/925.5 |
6.0 |
○ |
|
| A187AFTECCQAADKAACLLPK205 |
2125.0/2123.0 |
6.1 |
|
○ |
| V265HTECCHGDLLECADDRADLAK286 |
2585.1/2583.1 |
4.5 |
○ |
|
| Y287ICENQDSISSK298 |
1443.7/1441.7 |
4.4 |
|
○ |
| R361HPDYSVVLLLR372 |
1467.8/1465.8 |
8.8 |
○ |
○ |
| V397FDEFKPLVEEPQNLIK413 |
2045.1/2043.1 |
4.4 |
○ |
○ |
| Q414NCELFEQLGEYK426 |
1657.8/1655.8 |
4.3 |
|
○ |
| F427QNALLVR434 |
960.6/958.6 |
9.7 |
○ |
|
| K438VPQVSTPTLVEVSR452 |
1639.9/1637.9 |
8.7 |
○ |
○ |
| R509PCFSALEVDETYVPK524 |
1910.9/1908.9 |
4.7 |
○ |
○ |
| A570VMDDFAAFVEK581 |
1342.6/1340.6 |
4.0 |
|
○ |
| Transferrin (10 fmol) |
S47VIPSDGPSVACVK60 |
1415.7/1413.7 |
5.6 |
○ |
|
19.3 |
21.5 |
30.8 |
| K61ASYLDCIR69 |
1125.6/1123.6 |
8.2 |
○ |
|
| A62SYLDCIR69 |
997.5/995.5 |
5.9 |
○ |
|
| K122DSGFQMNQLR132 |
1323.6/1321.6 |
8.7 |
|
○ |
| C213LKDGAGDVAFVK225 |
1379.7/1377.9 |
6.0 |
○ |
○ |
| M331YLGYEYVTAIR343 |
1478.7/1476.7 |
5.8 |
○ |
○ |
| S453ASDLTWDNLK464 |
1249.6/1247.6 |
4.2 |
○ |
○ |
| F495DEFFSEGCAPGSK508 |
1577.7/1575.7 |
4.1 |
|
○ |
| F495DEFFSEGCAPGSKK509 |
1705.7/1703.7 |
4.7 |
○ |
○ |
| E531GYYGYTGAFR541 |
1283.6/1281.6 |
6.1 |
○ |
|
| C542LVEKGDVAFVK553 |
1364.7/1362.7 |
6.1 |
|
○ |
| H554QTVPQNTGGKNPDPWAK571 |
1975.0/1973.0 |
8.6 |
|
○ |
| N572LNEKDYELLCLDGTR587 |
1952.9/1950.9 |
4.3 |
○ |
○ |
| D647LLFRDDTVCLAK659 |
1565.8/1563.8 |
4.4 |
○ |
○ |
| Y669LGEEYVK676 |
1000.5/998.5 |
4.5 |
○ |
|
| Lysozyme (50 fmol) |
V20FGRCELAAAMK31 |
1368.7/1366.7 |
8.2 |
|
○ |
45.3 |
54.1 |
54.7 |
| G40YSLGNWVCAAK51 |
1325.6/1323.6 |
8.2 |
○ |
○ |
| F52ESNFNTQATNR63 |
1428.7/1426.7 |
6.0 |
○ |
○ |
| N64TDGSTDYGILQINSR79 |
1753.9/1751.9 |
4.2 |
○ |
○ |
| W80WCNDGR86 |
991.4/989.4 |
5.8 |
○ |
○ |
| C133KGTDVQAWIR143 |
1333.7/1331.7 |
8.2 |
○ |
|
| G135TDVQAWIR143 |
1045.5/1043.5 |
5.8 |
○ |
○ |
| α-Casein (50 fmol) |
H23QGLPQEVLNENLLR37 |
1760.0/1758.0 |
5.4 |
|
○ |
27.8 |
26.2 |
41.4 |
| E50KVNELS*K57 |
946.6/944.6 |
6.2 |
○ |
|
| D58IGS*ES*TEDQAMEDIK73 |
1943.9/1941.9 |
3.7 |
○ |
|
| H95IQKEDVPSER105 |
1337.8/1335.8 |
5.4 |
○ |
○ |
| E99DVPSERYLGYLEQLLR115 |
2080.1/2078.1 |
4.4 |
|
○ |
| Y106LGYLEQLLR115 |
1267.8/1265.8 |
6.0 |
|
○ |
| Y119KVPQLEIVPNS*AEER134 |
1952.0/1950.0 |
4.8 |
|
○ |
| V121PQLEIVPNS*AEER134 |
1660.9/1658.9 |
4.3 |
○ |
○ |
| T209TMPLW214 |
748.4/746.4 |
5.2 |
○ |
|
| T209TM*PLW214 |
764.4/762.4 |
5.2 |
○ |
|
Avoidance of false positive identifications
To validate false positive identifications, HSA, transferrin or α-casein was analyzed as unknown protein. The search results were listed until rank 5 proteins identified (Table 2). All the proteins were identified as rank 1 with good scores (>20) by both positive and negative ionizations. As we expected, none of the rank 2–5 proteins (false positive identifications) was overlapped between positive and negative ionizations. Therefore, comparison between positive and negative ionizations would be the way to eliminate false positive identifications.
Table 2 Top 5 proteins identified from HSA (100 fmol), transferrin (50 fmol), or α-casein (100 fmol) standard. Enzyme, trypsin; Missed cleavage, 1; Resolution, Monoisotopic; Modifications, carbamidomethyl (C), oxidation (M), phosphorylation (S, T and Y); Spectrometer shift max, ±0.2 Da; Spectrometer slope max, ±200 ppm; Spectrometer internal error max, ±25 ppm; Minimum number of hits, 4. Threshold: species, H. sapiens; Mw range, 60000–70000; pI range, 5–7 (for HSA); species, H. sapiens; Mw range, 60000–80000; pI range 5–7, (for transferrin); species, B. taurus; Mw range, 20000–40000; pI range, 4–7 (for α-casein)
| Proteins |
Ionization |
Rank |
Score |
Hits |
AC |
ID |
DE |
M
w/kDa |
pI |
Coverage (%) |
| HSA (100 fmol) |
Positive |
1 |
136.17 |
23 |
P02768 |
ALBU_HUMAN (C_1) |
Serum albumin |
66 |
5.7 |
50 |
| 2 |
6.25 |
9 |
Q96AY2 |
EME1_HUMAN (C_1) |
Crossover junction endonuclease EME1 |
63 |
6.7 |
17 |
| 3 |
4.17 |
7 |
Q96G03 |
PGM2_HUMAN (C_1) |
Phosphoglucomutase-2 |
68 |
6.3 |
16 |
| 4 |
3.64 |
7 |
Q9H9A6 |
LRC40_HUMAN (C_1) |
Leucin-rich repeat-containing protein 4… |
68 |
6.0 |
12 |
| 5 |
3.10 |
7 |
Q95391 |
SLU7_HUMAN (C_1) |
Pre-mRNA-splicing factor SLU7 |
68 |
6.7 |
13 |
| Negative |
1 |
131.07 |
19 |
P02768 |
ALBU_HUMAN (C_1) |
Serum albumin |
66 |
5.7 |
45 |
| 2 |
2.71 |
6 |
P38646 |
GRP75_HUMAN (C_1) |
Stress-70 protein. Mitochondrial |
69 |
5.5 |
15 |
| 3 |
2.52 |
5 |
Q14124 |
PGM5_HUMAN (C_1) |
Phosphoglucomutase-like protein 5 |
62 |
6.8 |
14 |
| 4 |
2.45 |
4 |
Q96A23 |
CPNE4_HUMAN (C_1) |
Copine-4 |
62 |
5.9 |
11 |
| 5 |
2.42 |
6 |
Q8WZ60 |
KLHL6_HUMAN (C_1) |
Kelch-like protein 6 |
69 |
5.8 |
8 |
| Transferrin (50 fmol) |
Positive |
1 |
135.19 |
24 |
P02787 |
TRFE_HUMAN (C_1) |
Serotransferrin |
75 |
6.7 |
35 |
| 2 |
5.49 |
7 |
P02545-2 |
LMNA_HUMAN (V_2) |
Isoform C of Lamin-A/C OS = H. sapiens G… |
65 |
6.4 |
11 |
| 3 |
4.85 |
7 |
Q86YJ7 |
AN13B_HUMAN (C_1) |
Ankyrin repeat domain-containing protein… |
70 |
6.5 |
15 |
| 4 |
4.73 |
7 |
P02545 |
LMNA_HUMAN (C_1) |
Lamin-A/C |
74 |
6.6 |
9 |
| 5 |
4.15 |
6 |
Q6L9W6-2 |
B4GN3_HUMAN (V_2) |
Isoform2 of N-acetyl-β-glucosaminyl-… |
63 |
6.8 |
12 |
| Negative |
1 |
33.39 |
14 |
P02787 |
TRFE_HUMAN (C_1) |
Serotransferrin |
75 |
6.7 |
23 |
| 2 |
3.00 |
7 |
Q81UR7 |
ARMC8_HUMAN (C_1) |
Armadillo repeat-containing protein 8 |
75 |
6.3 |
10 |
| 3 |
2.83 |
7 |
Q81UR7-3 |
ARMC8_HUMAN (V_3) |
Isoform 3 of Armadillo repeat-containing… |
71 |
6.4 |
9 |
| 4 |
2.71 |
7 |
Q81UR7-2 |
ARMC8_HUMAN (V_2) |
Isoform 2 of Armadillo repeat-containing… |
74 |
6.3 |
9 |
| 5 |
2.08 |
5 |
Q8WYL5-2 |
SSH1_HUMAN (V_2) |
Isoform 2 of Protein phosphatase Slingsh… |
77 |
5.8 |
9 |
| α-Casein (100 fmol) |
Positive |
1 |
20.39 |
9 |
P02662 |
CASA1_BOVIN (C_1) |
α-S1-Casein |
23 |
4.9 |
38 |
| 2 |
6.79 |
7 |
Q08E20 |
ESTD_BOVIN (C_1) |
S-Formylglutathione hydrolase |
32 |
6.5 |
29 |
| 3 |
5.14 |
6 |
Q3T0X7 |
NSE1_BOVIN (C_1) |
Non-structural maintenance of chromosome… |
31 |
6.6 |
19 |
| 4 |
4.79 |
6 |
A5PJP6-2 |
BRCC3_BOVIN (V_2) |
Isoform 2 of Lys-63-specific deubiquitin… |
33 |
5.6 |
18 |
| 5 |
4.49 |
5 |
P85100 |
MYL3_BOVIN (C_1) |
Myosin light chain 3 |
22 |
5.0 |
33 |
| Negative |
1 |
31.54 |
8 |
P02662 |
CASA1_BOVIN (C_1) |
α-S1-Casein |
23 |
4.9 |
41 |
| 2 |
6.34 |
5 |
P09611 |
CSH1_BOVIN (C_1) |
Chorionic somatomammotropin hormone 1 |
23 |
6.4 |
25 |
| 3 |
1.97 |
4 |
Q1RMX9 |
DCNL5_BOVIN (C_1) |
DCN1-like protein 5 |
27 |
5.6 |
25 |
| 4 |
1.41 |
5 |
Q2YDC9 |
PDCD2_BOVIN (C_1) |
Programmed cell death protein 2 |
39 |
5.4 |
11 |
| 5 |
1.16 |
4 |
Q2KI14 |
ARD1A_BOVIN (C_1) |
N-terminal acetyltransferase complex ARD… |
27 |
5.4 |
7 |
Conclusions
In summary, we have optimized the robust conditions to ionize intact peptides for negative ionization mode MALDI. Interestingly, the removal of alkali metals was more effective in ionizing peptides negatively rather than the basic atmosphere. Using these conditions, we have also demonstrated that the concomitant use of positive and negative ionizations for PMF can simply provide more reliable protein identification with higher peptide coverage and less false positive identifications. Because this strategy is simple, practical, reproducible and more effective for lower concentration samples, it has already been applied for the identification of proteins to avoid false positive identifications from complicated samples by finding common proteins identified from both positive and negative ion modes.
Acknowledgements
The authors thank Biomedical Research Core, School of Medicine in our University for the use of MALDI-TOFMS.
References
- M. Yamashita and J. B. Fenn, J. Phys. Chem., 1984, 88, 4451 CrossRef CAS.
- K. Tanaka, H. Waki, Y. Ido, S. Akita, Y. Yoshida, T. Yoshida and T. Matsuo, Rapid Commun. Mass Spectrom., 1988, 2, 151 CAS.
- A. Persidis, Nat. Biotechnol., 1998, 16, 393 CAS.
- V. Brower, EMBO Rep., 2001, 2, 558 CrossRef CAS.
- B. Thiede, W. Höhenwarter, A. Krah, J. Mattow, M. Schmid, F. Schmidt and P. R. Jungblut, Methods, 2005, 35, 237 CrossRef CAS.
- M. C. Fitzgerald, G. R. Parr and L. M. Smith, Anal. Chem., 1993, 65, 3204 CrossRef CAS.
- R. L. Vermillion-Salsbury and D. M. Hercules, Rapid Commun. Mass Spectrom., 2002, 16, 1575 CrossRef CAS.
- R. Shroff, A. Muck and A. Svatoš, Rapid Commun. Mass Spectrom., 2007, 21, 3295 CrossRef CAS.
- H. Nonami, K. Tanaka, Y. Fukuyama and R. Erra-Balsells, Rapid Commun. Mass Spectrom., 1998, 12, 285 CrossRef CAS.
- H. Nonami, F. Wu, R. P. Thummel, Y. Fukuyama, H. Yamaoka and R. Erra-Balsells, Rapid Commun. Mass Spectrom., 2001, 15, 2354 CrossRef CAS.
- I. P. Smirnov, X. Zhu, T. Taylor, Y. Huang, P. Ross, I. A. Papayanopoulos, S. A. Martin and D. J. Pappin, Anal. Chem., 2004, 76, 2958 CrossRef CAS.
- J. M. Asara and J. Allison, Anal. Chem., 1999, 71, 2866 CrossRef CAS.
- M. F. Weng and Y. C. Chen, Rapid Commun. Mass Spectrom., 2004, 18, 1421 CrossRef CAS.
- J. M. Asara and J. Allison, J. Am. Soc. Mass Spectrom., 1999, 10, 35 CrossRef CAS.
- J. H. Kang, R. Toita, J. Oishi, T. Niidome and Y. Katayama, J. Am. Soc. Mass Spectrom., 2007, 18, 1925 CrossRef CAS.
- S. Kjellström and O. N. Jensen, Anal. Chem., 2004, 76, 5109 CrossRef.
- A. Tholey, Rapid Commun. Mass Spectrom., 2006, 20, 1761 CrossRef CAS.
- H. Kuyama, K. Sonomura and O. Nishimura, Rapid Commun. Mass Spectrom., 2008, 22, 1109 CrossRef CAS.
- K. Janek, H. Wenschuh, M. Bienert and E. Krause, Rapid Commun. Mass Spectrom., 2001, 15, 1593 CrossRef CAS.
- T. Nishikaze and M. Takayama, Rapid Commun. Mass Spectrom., 2007, 21, 3345 CrossRef CAS.
- J. Gao and C. J. Cassady, Rapid Commun. Mass Spectrom., 2008, 22, 4066 CrossRef CAS.
- T. Nabetani, K. Miyazaki, Y. Tabuse and A. Tsugita, Proteomics, 2006, 6, 4456 CrossRef CAS.
- P. M. Bigwarfe, Jr and T. D. Wood, Int. J. Mass Spectrom., 2004, 234, 185 Search PubMed.
- S. T. Tsai, C. W. Chen, L. C. L. Huang, M. C. Huang, C. H. Chen and Y. S. Wang, Anal. Chem., 2006, 78, 7729 CrossRef CAS.
Footnotes |
| † These authors contributed equally to this work. |
| ‡ Present name and address: Mao Maekawa, Department of Pharmaceutical Sciences, Tohoku University Hospital, 1-1 Seiryo-machi, Aoba-ku, Sendai 980-8574, Japan. |
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.