An air-assisted liquid–liquid extraction using a dual-valve sequential injection manifold (DV-SIA): Determination of copper
Jana
Škrlíková
*a,
Vasil
Andruch
*a,
Hana
Sklenářová
*b,
Petr
Chocholouš
b,
Petr
Solich
b and
Ioseph S.
Balogh
c
aDepartment of Analytical Chemistry, University of P.J. Šafárik, SK-04154, Košice, Slovak Republic. E-mail: jana.skrlikova@googlemail.com; vasil.andruch@gmail.com
bDepartment of Analytical Chemistry, Faculty of Pharmacy, Charles University, CZ-50005, Hradec Králové, Czech Republic. E-mail: Hana.Sklenarova@faf.cuni.cz
cDepartment of Chemistry, College of Nyíregyháza, HU-4400, Nyíregyháza, Hungary
First published on 17th June 2010
Abstract
This work introduces an approach to liquid–liquid extraction for the sequential injection technique by improving on a previously reported dual-valve sequential injection manifold (DV-SIA). The system is made up of three units: a Mixing unit (for aqueous phase only), an Extraction unit, and a Detection unit (for organic phase only). The extraction was carried out by controlled aeration in the Extraction unit. The efficiency of the suggested extraction procedure was demonstrated by the spectrophotometric determination of copper extracted as an ion associate of Cu(I) with the polymethine dye 1,3,3-trimethyl-2-[5-(1,3,3-trimethyl-1,3-dihydroindol-2-ylidene)-penta-1,3-dienyl]-3H-indolium (DIDC). Appropriate experimental conditions were found to be: 40 μL of 2 mol L−1 NaCl; 50 μL of sample, 20 μL of 1 mmol L−1 DIDC (pH 3), flow rate 50 μL s−1. A linear response was obtained in the range 0.13–2.0 μg mL−1 of Cu, and the limit of detection, calculated based on three times the standard deviation of the blank test (n = 10), was 0.02 μg mL−1. The method was applied to an assay of copper in pharmaceutical materials.
Introduction
Air (or inert gas) aeration has been frequently used in a variety of chemical technologies, mostly in order to improve mixing. However, its application in analytical chemistry is less widely used. We found a few interesting examples in the literature: Dasgupta et al. used air bubbling to overcome the interference of high manganese content based on the oxidation of Mn(II)–TEA to Mn(III)–TEA during the titration of calcium at pH 12;1 Idowu et al. used ambient air to generate ozone in a gas-phase chemiluminescence-based analyser for waterborne arsenic;2 Pereira et al. reported methods for the determination of Mn and Ni in lake and marine sediment slurries by GFAAS, in which slurries were kept homogeneous by air bubbling with an aquarium pump;3 Camou et al. described a portable sensor for measuring benzene in water based on bubbling extraction and UV spectroscopy detection.4Air has also been widely used in flow-based systems for segmentation or as a carrier. Jakmunee et al. used air to minimise the dispersion between the sample-reagent zone and the carrier stream;5 Leelasattarathkul et al. used it to promote the mixing efficiency of sample and reagents;6 Wang and Hansen used air to transport analyte into the graphite tube of an electrothermal atomic absorption spectrometer after separation/preconcentration of analyte by various methods;7–11 Hong-Bing et al. employed air to form a barrier between sample and reagent in order to prevent a reaction during the aspiration stage;12 and Stefanova et al. used it to isolate the sample from the water carrier.13
Probably the oldest and most widely investigated separation/preconcentration technique is liquid–liquid extraction (LLE). However, manually performing this technique has numerous limitations and drawbacks which are well known. Therefore in the second half of the 20th century, so-called solvent-free sample preparation techniques began to be developed, such as sorbent extraction. Here it is necessary to underline the works of Pawliszyn in particular.14–16 Recently, however, interest in the use of LLE has been revived due to its advantages, and novel approaches using LLE techniques that require only a small amount of solvent have been suggested,17 especially in automated forms. The incorporation of LLE into the more versatile second generation flow system called sequential injection analysis (SIA) seems to be a feasible way for the wider application of LLE in analytical practice. Although many articles have dealt with FIA-LLE systems, it is worth noting that only a few have dealt with the SIA-LLE approach.
Copper is an essential trace element which can be vital or toxic to biological systems, depending on the level of concentration.18 A variety of techniques for copper determination exist, and the analytical chemistry of copper has been discussed in numerous books19,20 and reviews.21–25 Spectrophotometric detection continues to be widely popular due to its simplicity and the availability of instrumentation. Dithiocarbamate, 1,10-phenanthroline and dithizone are the most widespread reagents for copper determination.19,20
Recently, a few interesting articles devoted to copper determination have been published: Amin reported a procedure for the determination of trace amounts of copper based on solid-phase spectrophotometry;26 Liang et al. described near-infrared-emitting CdSeTe alloyed quantum dots capped with L-cysteine for ultrasensitive Cu2+ sensing;27 Anthemidis et al. developed an on-line sequential injection dispersive liquid–liquid microextraction system for flame atomic absorption spectrometric determination of copper and lead in water samples;28 Rumori and Cerda compared FIA and SIA for the spectrophotometric determination of Cu(II) in water at trace levels based on the reaction with cuprizone in alkaline media;29 van Staden and Taljaard suggested an instrumental system for simultaneous determination of seven different metal ions using dithizone in ethanol as extractant and sequential injection thin-film extraction based on the hydrophobic interaction of ethanol with the Teflon wall to create a thin film;30 Ohno et al. reported a striking SI method in a lab-on-valve format for simultaneous spectrophotometric determination of copper and iron based on the complex formation of 2-(5-bromo-2-pyridylazo)-5-[N-n-propyl-N-(3-sulfopropyl)amino]aniline with Cu(II) and/or Fe(II);31 Vidotti et al. described a procedure involving the bead-injection concept and sequential determination of copper and mercury ions in river water samples based on the solid-phase extraction of both metal ions on the same bead's surface and on their subsequent reaction with the colorimetric reagents.32
We previously reported a novel sequential injection system based on a dual-valve (DV-SIA)33,34 approach for online liquid–liquid extraction which was constructed by the connection of two independent units, one for aqueous-organic mixture flow (the so-called Extraction unit) and the second specifically for organic phase flow (the so-called Detection unit). Aspirating only organic phase into the Detection unit circumvented some of the problems caused by the different affinities of aqueous and organic phase to the walls of the Teflon tubing used in the SI-system. However, this design did not overcome all the difficulties associated with the incorporation of LLE into an SIA manifold, because in the Extraction unit both aqueous and organic phases were still used.
This work introduces an air-assisted approach to liquid–liquid extraction for a previously reported dual-valve sequential injection (DV-SIA) manifold. The improved system is made up of three units: a Mixing unit (for aqueous phase only), an Extraction unit, and a Detection unit (for organic phase only). The feasibility of the suggested system was demonstrated for the extractive-spectrophotometric determination of copper in the form of ion associate of Cu(I) with polymethine dye 1,3,3-trimethyl-2-[5-(1,3,3-trimethyl-1,3-dihydro-indol-2-ylidene)-penta-1,3-dienyl]-3H-indolium (DIDC).
Experimental
Reagents
All solutions used were prepared from analytical grade reagents, and the organic solvents (amyl acetate, methanol) were of UV spectroscopy grade. Ultra pure water from Millipore Milli-Q RG (Millipore, USA) was used throughout the work. The working solution containing 1.2 μg mL−1 Cu(I) was prepared daily from Cu(II) stock solution (1000 mg L−1) by appropriate step-wise dilution and simultaneous reduction by addition of a 2% solution of ascorbic acid. A 2 mol L−1 aqueous solution of chloride ions (NaCl) was used as ligand. A 1 mmol L−1 solution of dye reagent was prepared by dissolving 0.0209 g of 1,3,3-trimethyl-2-[5-(1,3,3-trimethyl-1,3-dihydro-indol-2-ylidene)-penta-1,3-dienyl]-3H-indolium chloride (DIDC) in 0.5 mL of methanol, followed by the addition of water, adjustment of the pH to 3 by the addition of 5 mL 0.01 mol L−1 HCl, and completing to a volume of 50 mL with water.Apparatus
The sequential injection system (Fig. 1) was constructed from following parts: (1) Mixing unit: FIAlab® 3500 system (FIAlab® Instrument Systems Inc., Dunedin, FL, USA) with a 5 mL syringe pump, 0.75 mm i.d. PTFE tubing 150 cm long (for the aspiration of water as the carrier, 1.5 mm i.d. PTFE tubing was used) and an 8-port Cheminert selection valve; (2) Extraction unit: A small unit from polypropylene with a volume of 1.5 mL closed by a top with four small holes for the tubes used for the input/output of aqueous phase, organic solvent, air and the aspiration of organic phase after extraction; (3) Detection unit: An external 6-port Cheminert selection valve (Valco Instrument Co., Houston, USA), 0.75 mm i.d. PTFE tubing 34 cm long and an external 5 mL syringe pump (FIAlab® Instrument Systems Inc., Dunedin, FL, USA). This SIA set-up was supplemented with a VIS light source LS-1 tungsten lamp and with a fibre-optic charge-coupled detector USB 2000 (both Ocean Optics Inc., Dunedin, FL, USA) and a microvolume Z-flow cell of 20 mm optical path length. All parts of the DV-SIA system were controlled by the latest version of the FIALab® software.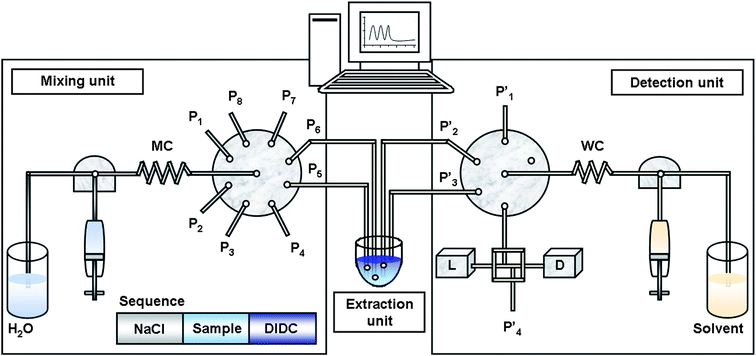 | ||
Fig. 1 Schematic view of the DV-SIA system for air-assisted liquid–liquid extraction. P1, P1′ – auxiliary wastes; P2 – NaCl; P3 – Cu(I)/sample; P4 – DIDC (pH 3); P5 – emptying of the Mixing unit into the Extraction unit; P6 – air output; P7 – air input; P8 – MeOH–H2O (v:v/4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1); P2′ – amyl acetate; P3′ – aspiration of organic phase after the extraction; P4′ – waste; L – VIS light source; D – charge-coupled detector; MC – mixing coil (150 cm length, 0.75 mm i.d.); WC – waiting coil (34 cm length, 0.75 mm i.d.) :1); P2′ – amyl acetate; P3′ – aspiration of organic phase after the extraction; P4′ – waste; L – VIS light source; D – charge-coupled detector; MC – mixing coil (150 cm length, 0.75 mm i.d.); WC – waiting coil (34 cm length, 0.75 mm i.d.) | ||
All aqueous solutions as well as the water used as a carrier were degassed using a Sonorex RK 100 ultrasonic bath (Bandelin Electronic, Berlin, Germany) before the sequential injection analysis.
General SIA procedure
The 500 μL of water as carrier was aspirated at a flow rate of 100 μL s−1 into the reservoir of the syringe pump of the Mixing unit. Then, 40 μL of 2 mol L−1 NaCl (P2), 50 μL of sample containing copper in the form Cu(I) (P3) and 20 μL of 1 mmol L−1 DIDC (P4) were aspirated into the mixing coil at a flow rate of 50 μL s−1. The aspirated zones were subsequently dispensed (P5) into the Extraction unit. In the next step, 300 μL of amyl acetate as extraction solvent was dispensed (P2′) into the Extraction unit from the reservoir of the Detection unit syringe pump. For extraction, 700 μL of air aspirated before via the Mixing unit (P7), was emptied (P6) into the middle of Extraction unit at a flow rate 50 μL s−1. After the mixing by air, the phases were self-separated based on their different densities. Next, 100 μL of organic phase was aspirated (P3′) into the waiting coil of the Detection unit and subsequently transferred to the detector using the spare 300 μL of amyl acetate in the syringe pump reservoir. The absorbance of the formed ion associate (see below) was measured in a 20-mm Z-flow cell at wavelengths of 640 nm.After each measurement, the Mixing unit and Extraction unit were cleaned using a water–methanol mixture (v:v/1:4).
Results and discussion
The method is based on the formation of ion associate between copper(I) and DIDC with the aid of chloride ions as ligand, which is extractable by the organic solvents.35 The reaction mechanism for the formation and extraction of ion associate may be expressed by the scheme:35| Cu(aq)+ + 2Cl(aq)− + R(aq)+ + nS(org) = [CuCl2]−R+ × nS(org) |
Investigation of appropriate experimental conditions
Despite the fact that the experimental conditions for Cu(I)-Cl-DIDC were determined and described in our laboratory previously,35 it was necessary to specify these conditions again in flow mode. The chemical conditions (pH, concentration of NaCl, and DIDC) as well as physical conditions (reagents volume, flow rate, the sequence order) were therefore studied. The main criteria to be obtained were: (1) the highest analytical signal; (2) the lowest value of the blank; and (3) simultaneously achieve the best possible precision. Three repetitions of all measurements were performed.The effect of the acidity of the medium was investigated in the range of pH 1–5 (Fig. 2), and pH 3 was chosen throughout the further study. The concentration of the reagents was investigated in the range 0.5–3 mol L−1 of NaCl and 0.2–1 mmol L−1 of DIDC (Fig. 3), and the concentrations 2 mol L−1 NaCl and 1 mmol L−1 DIDC were selected. The volume of the reagents (Fig. 4) was investigated in 10 μL increments (the volume of sample held constant at 50 μL), and 40 μL of NaCl and 20 μL of DIDC were chosen for further study.
 | ||
| Fig. 2 Investigation of the acidity of the medium 40 μL of 2 mol L−1 NaCl; 50 μL of 1.2 μg mL−1 Cu(I); 20 μL of 1 mmol L−1 DIDC; λ = 640 nm; 20 mm Z-flow cell; 300 μL amyl acetate. | ||
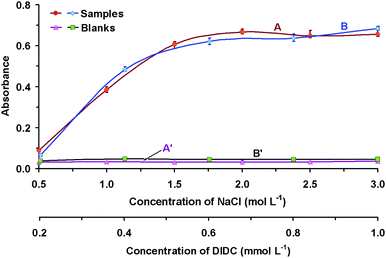 | ||
| Fig. 3 Investigation of reagent concentrations. Effect of the concentration of NaCl (A, A′) and DIDC (B, B′); 50 μL of 1.2 μg mL−1 Cu(I); λ = 640 nm; 20 mm Z-flow cell; 300 μL amyl acetate; A, B – ion associates, A′, B′ – blank tests; A, A′ – 40 μL NaCl; 20 μL of 1 mmol L−1 DIDC; B, B′ – 40 μL of 2 mol L−1 NaCl; 20 μL DIDC | ||
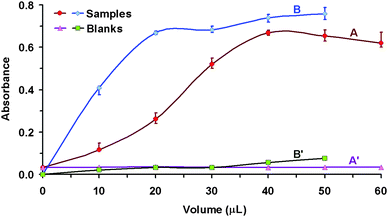 | ||
| Fig. 4 Investigation of reagent volumes. Effect of the volume of NaCl (A, A′) and DIDC (B, B′); λ = 640 nm; 20 mm Z-flow cell; 50 μL of 1.2 μg mL−1 Cu(I); 300 μL amyl acetate; A, B – ion associates, A′, B′ – blank tests; A, A′ – 2 mol L−1 NaCl; 20 μL of 1 mmol L−1 DIDC; B, B′ – 40 μL of 2 mol L−1 NaCl; 1 mmol L−1 DIDC. | ||
The formation of ion-associate is fast; therefore, the flow rate of aspiration of the reagents was not so crucial. However, the reagents and sample must be mixed well, and this was assured during emptying of the Mixing unit into the Extraction unit. The flow rate of 50 μL s−1 was chosen for further experiments.
The sequence of the aspiration of the sample and the reagents proved to be at least as important as the concentration or volume of the reagents. Various sequences were examined, such as NaCl–sample–DIDC; sample–NaCl–DIDC; DIDC–NaCl–sample; DIDC–sample–NaCl–DIDC and NaCl–sample–DIDC–NaCl. The highest analytical signal with the best precision was obtained with two sequences: NaCl–sample–DIDC and DIDC–sample–NaCl–DIDC. Due to the shorter SIA procedure time, the sequence NaCl–sample–DIDC was chosen.
Investigation of appropriate extraction conditions
The following parameters were studied: (1) amount of air needed for extraction; (2) air flow rate; (3) volume of organic solvent. If necessary due to the reaction mechanism, the air can be replaced by an inert gas, such as nitrogen. However, it complicates the whole procedure and makes it more expensive. Despite the fact that Cu(I) has reductive propertives, the presence of an abundance of ascorbic acid in the solution prevents oxidation to Cu(II). Therefore, in our case, air was selected for further experiments. The amount of air was investigated in the range 100–1000 μL at a flow rate of 50 μL s−1. The analytical signal was increased up to 600 μL, and the best values of RSD were obtained in the range 700–1000 μL. Therefore, the amount 700 μL air was chosen due to the shorter analysis time. The flow rate of air bubbling was studied in the range 20–70 μL s−1. However, no significant changes were found, so a flow rate of 50 μL s−1 was selected. Based on previous work,35 amyl acetate was used as the extraction solvent. The minimum volume of amyl acetate for extraction was 300 μL. When less than 300 μL of organic solvent was used, it was difficult to achieve complete separation between the two phases, because the aqueous droplets were caught in the organic phase.Method validation
A calibration plot was constructed using solutions with different quantities of copper prepared from standard reference material (Scharlau CO 00850500, UN 3264, 1000 mg L−1) by appropriate step-wise dilution and simultaneous reduction using a 2% solution of ascorbic acid. A linear analytical signal was obtained in the range 0.13–2.0 μg mL−1 of Cu with the regression equation A = 0.0037 + 0.4726 × C (where A means the absorbance, and C the concentration of Cu in μg mL−1) and with a correlation coefficient 0.9992. The limit of detection (LOD), calculated based on three times of the standard deviation of the blank test (n = 10), was 0.02 μg mL−1. The precision (as relative standard deviation, RSD%) and accuracy (as recovery percentage, R%) of the method were examined by performing 10 determinations at three concentration levels (0.4; 0.8; 1.4 μg mL−1) over two days (Table 1).| Taken/μg mL−1 | Intra-day | Inter-day | ||||
|---|---|---|---|---|---|---|
| Determineda/μg mL−1 | RSD (%) | R (%) | Determineda/μg mL−1 | RSD (%) | R (%) | |
a
 (t = 2.262, P = 0.95); t – Student coefficient for n–1 degrees of freedom. (t = 2.262, P = 0.95); t – Student coefficient for n–1 degrees of freedom.
|
||||||
| 0.40 | 0.41 ± 0.01 | 3.4 | 102.5 | 0.39 ± 0.01 | 3.6 | 97.5 |
| 0.80 | 0.79 ± 0.01 | 1.8 | 98.8 | 0.81 ± 0.01 | 1.7 | 101.3 |
| 1.40 | 1.40 ± 0.02 | 2.0 | 100.0 | 1.39 ± 0.02 | 2.0 | 99.3 |
Selectivity
The effect of some interfering ions on the determination of 0.66 μg mL−1 of Cu(I) was examined. A ratio of Cu:interferent which resulted in an error not exceeding ± 5% was taken as the tolerable amount of each ion. Most of the examined ions (Cd2+, Pb2+, Zn2+, Fe3+, Ag+, Mg2+, Ca2+) did not interfere with the determination of copper at more than a 100-fold excess; Co2+, Ni2+ and Al3+ did not interfere at more than a 10-fold excess; the anions (CH3COO−, SO42−, NO3−) did not disturb determination at more than a 2000-fold excess.The matrix effect was studied by spiking commercially available mineral water (Ca2+ (83.9); Mg2+ (19); Na+ (<1.5); K+ (0.6); NH4+ (0.04); HCO3− (210); SO42− (19.4); NO3− (11.4); Cl− (3.0); F− (0.1); CO2 (<0.1); NO2− (<0.01) mg L−1) and a pharmaceutical preparative containing 500 mg of paracetamol per tablet and excipients, with copper solution at different concentration levels and analysing them using the suggested procedure. The pharmaceutical preparative was pre-treated before analysis: one tablet was crushed and treated in a flask with small amounts of water using ultrasound for 40 min. Next, the undissolved excipients were filtered out, and the filtrate was gathered into a 50 mL volumetric flask and filled up with water. An aliquot portion of solution prepared in this manner was applied as a matrix in the determination of copper. The results given in Table 2 showed good precision and accuracy of the determination with no matrix interference.
| Matrix | Added/μg mL−1 | Determineda/μg mL−1 | RSD (%) | R (%) |
|---|---|---|---|---|
| a (t = 2.262, P = 0.95). b in the presence of 25 μg mL−1 of paracetamol. | ||||
| Mineral water | 0.50 | 0.49 ± 0.01 | 2.9 | 98.0 |
| 0.90 | 0.90 ± 0.01 | 1.6 | 100.0 | |
| 1.50 | 1.49 ± 0.02 | 1.9 | 99.3 | |
| Pharmaceutical preparative | 0.80b | 0.79 ± 0.02 | 3.5 | 98.8 |
| 1.40b | 1.41 ± 0.02 | 2.0 | 100.7 |
Analytical application
To demonstrate the practicability of the suggested DV-SIA system using aeration for liquid–liquid extraction, the technique was applied to the determination of copper in real pharmaceutical samples containing various amount of copper (0.75 mg and 2.5 mg Cu per tablet). The sample was pre-treated prior to analysis: one tablet was dissolved (ultrasonicated for 60 min, if needed), filtered and filled to the volume of 100 mL with water. A 2% solution of ascorbic acid was added to various aliquots of this prepared solution; the aliquots were then completed with water and used for the subsequent analysis. The obtained results (Table 3) show sufficient agreement between the taken and determined values.| Content of pharmaceuticals (mg per tablet) | Taken/μg mL−1 | Added/μg mL−1 | Determineda/μg mL−1 | RSD (%) | R (%) |
|---|---|---|---|---|---|
| a (t = 2.262, P = 0.95). | |||||
| Ca (75.0), Mg (30.0), P (57.9), Fe (3.0), Zn (3.0), Mn (0.75), I (0.045), K (15.0), Cl (13.5), Cr (0.0375), Mo (0.0375), Se (0.0375)), vitamins C, E and vitamins of B group, excipients | 0.38 | — | 0.37 ± 0.01 | 3.8 | 97.4 |
| 0.75 | — | 0.76 ± 0.01 | 1.8 | 101.3 | |
| 1.13 | — | 1.12 ± 0.01 | 1.2 | 99.1 | |
| 1.88 | — | 1.87 ± 0.02 | 1.5 | 99.5 | |
| Ca (162); I (0.15); Fe (18); P (126); Mg (100); Zn (15); Se (0.02); Cu (2.5); Mn (2.5); Cr (0.025); Mo (0.025); Cl (36.3); K (39), vitamins A, C, D, E, K, and vitamins of B group, excipients | 0.50 | 1.0 | 1.47 ± 0.04 | 3.8 | 98.0 |
| 0.80 | 0.48 | 1.30 ± 0.02 | 2.2 | 101.6 | |
Conclusions
Comparing the suggested sequential injection procedure for copper determination with the previously described conventional manual extraction procedure,35 both of which are based on the use of the same reagent as well as the same reaction mechanism, we can conclude that the novel DV-SIA manifold using aeration for extraction shows several advantages: (i) a closed system for the handling of organic solvent; (ii) lower volume consumption of organic solvent; (iii) production of a lower amount of organic waste; (iv) reduced risk of analyte loss and sample contamination; and (v) better precision and accuracy. The linear ranges and LODs are comparable for both procedures.The implementation of air bubbling into the DV-SIA system and employing of three independent units (one for aqueous phase only, one for organic phase only, and one for extraction using air bubbling), allowed for the complete separation of the aqueous and organic phases in the tubing of the system, thus preventing film formation and avoiding of the problem caused by disruption of the film by the change of organic and aqueous phases flowing through the system. Therefore, factors which can cause the appearance of a parasitic signal and consequently worsen the repeatability and reproducibility of the results were eliminated or minimised.
Incorporation of air-assisted extraction into a dual-valve sequential injection system demonstrates the flexibility of the previously reported DV-SIA manifold33 and also suggests possible use for other systems with markedly different demands for the extraction process.
Acknowledgements
The authors would like to say a special thanks to Professor Dr J. Ruzicka, USA, for the fruitful discussion. H.S. and P.S. would like to thank to the Ministry of Education of the Czech Republic for its financial support (MSM 0021620822). J.S. would like to say a special thanks to the International Visegrad Fund for providing a 10-month Scholarship and the internal grant system of Faculty of Science of Pavol Jozef Šafárik University in Košice (VVGS Grant No. 24/2009/CH).References
- S. Dasgupta, B. C. Sinha and N. S. Rawat, Analyst, 1983, 108, 1396–1401 RSC.
- A. D. Idowu, P. K. Dasgupta, Z. Genfa, K. Toda and J. R. Garbarino, Anal. Chem., 2006, 78, 7088–7097 CrossRef CAS.
- L. A. Pereira, S. S. O. De Borges, M. C. Castro, W. B. Neto, C. C. Windmöller and J. B. B. Da Silva, Can. J. Chem., 2008, 86, 312–316 CrossRef CAS.
- S. Camou, A. Shimizu, T. Horiuchi and T. Haga, Sens. Actuators, B, 2008, 132, 601–607 CrossRef.
- J. Jakmunee, L. Pathimapornlert, S. K. Hartwell and K. Grudpan, Analyst, 2005, 130, 299–303 RSC.
- T. Leelasattarathkul, S. Liawruangrath, M. Rayanakorn, W. Oungpipat and B. Liawruangrath, Talanta, 2006, 70, 656–660 CrossRef CAS.
- J. Wang and E. H. Hansen, Anal. Chim. Acta, 2002, 456, 283–292 CrossRef CAS.
- J. Wang and E. H. Hansen, J. Anal. At. Spectrom., 2002, 17, 248–252 RSC.
- J. Wang and E. H. Hansen, Anal. Chim. Acta, 2001, 435, 331–342 CrossRef CAS.
- J. Wang and E. H. Hansen, Atom. Spectrosc., 2001, 22, 312–318 CAS.
- J. Wang and E. H. Hansen, Anal. Chim. Acta, 2000, 424, 223–232 CrossRef CAS.
- M. Hong-Bing, Zh.-L. Fang, J.-F. Wu and Sh.-Sh. Liu, Talanta, 1999, 49, 125–133 CrossRef CAS.
- V. Stefanova, V. Kmetov and L. Futekov, J. Anal. At. Spectrom., 1997, 12, 1271–1276 RSC.
- C. L. Arthur and J. Pawliszyn, Anal. Chem., 1990, 62, 2145–2148 CrossRef CAS.
- S. Risticevic, V. H. Niri, D. Vuckovic and J. Pawliszyn, Anal. Bioanal. Chem., 2009, 393, 781–795 CrossRef CAS.
- G. Ouyang and J. Pawliszyn, Anal. Chim. Acta, 2008, 627, 184–197 CrossRef CAS.
- F. Pena-Pereira, I. Lavilla and C. Bendicho, Spectrochim. Acta, Part B, 2009, 64, 1–15 CrossRef.
- W. Kaim and B. Schwederski, Bioinorganic Chemistry. Inorganic Elements in the Chemistry of Life, John Wiley & Sons, New York, 1991, p. 187–212 Search PubMed.
- V. N. Podchinova and L. N. Simonova, in The Copper (in Russian), Nauka, Moscow, 1990, p. 92–120 Search PubMed.
- Z. Marczenko and M. Balcerzak, in Separation, Preconcentration and Spectrophotometry in Inargonic Analysis, Elsevier, Amsterdam, 1st edn, 2000, p. 177–188 Search PubMed.
- J. Sardans, F. Montes and J. Peñuelas, Spectrochim. Acta, Part B, 2010, 65, 97–112 CrossRef.
- G. Herzog and D. W. M. Arrigan, TrAC, Trends Anal. Chem., 2005, 24, 208–217 CrossRef CAS.
- P. Kuban, R. Guchardi and P. C. Hauser, TrAC, Trends Anal. Chem., 2005, 24, 192–198 CrossRef CAS.
- M. Miró, J. M. Estela and V. Cerdà, Talanta, 2004, 63, 201–223 CrossRef CAS.
- T. P. Rao, M. L. P. Reddy and A. R. Pillai, Talanta, 1998, 46, 765–813 CrossRef.
- A. S. Amin, Chem. Pap., 2009, 63, 625–634 Search PubMed.
- G.-X. Liang, H.-Y. Liu, J.-R. Zhang and J.-J. Zhu, Talanta, 2010, 80, 2172–2176 CrossRef CAS.
- A. N. Anthemidis and K.-I. G. Ioannou, Talanta, 2009, 79, 86–91 CrossRef CAS.
- P. Rumori and V. Cerdà, Anal. Chim. Acta, 2003, 486, 227–235 CrossRef CAS.
- J. F. van Staden and R. E. Taljaard, Talanta, 2004, 64, 1203–1212 CrossRef CAS.
- Sh. Ohno, N. Teshima, T. Sakai, K. Grudpan and M. Polasek, Talanta, 2006, 68, 527–534 CrossRef CAS.
- E. C. Vidotti, V. C. Almeida and C. C. Oliveira, Talanta, 2004, 64, 993–999 CrossRef CAS.
- J. Škrlíková, V. Andruch, H. Sklenářová, P. Chocholouš, P. Solich and I. S. Balogh, Anal. Chim. Acta, 2010, 666, 55–61 CrossRef CAS.
- A device for sequential injection analysis for liquid–liquid extraction, Czech Patent application No. PV 2009–726.
- I. S. Balogh, M. Ruschak, V. Andruch and Y. Bazel', Talanta, 2008, 76, 111–115 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |