Determination of dissolved oxygen based on photoinduced electron transfer from quantum dots to methyl viologen†
Cong
Kong
,
Lixia
Qin
,
Jiaofang
Liu
,
Xinhua
Zhong
,
Linyong
Zhu
* and
Yi-Tao
Long
*
Key Laboratory for Advanced Materials & Department of Chemistry, East China University of Science and Technology, 130 Meilong Road, Shanghai, P.R. China. E-mail: ytlong@ecust.edu.cn; linyongzhu@ecust.edu.cn; Tel: +86 21 642 50032 Tel: +86 21 642 52339
First published on 21st June 2010
Abstract
A rapid colorimetric methodology based on photoinduced electron transfer from excited CdS quantum dots (CdS QDs) to methyl viologen (MV2+) has been developed for the sensitive determination of dissolved oxygen (DO) content. The reduction of MV2+ is initiated by light excitation of CdS QDs, which induces the electron transfer from sacrificial donor glutathione (GSH) to the photogenerated hole of CdS QDs. Due to the presence of oxygen, the reduced radical-cation of methyl viologen (MV+˙) could be rapidly reoxidized, thus turning into the original methyl viologen dication (MV2+). The existence of MV+˙ is confirmed by electron paramagnetic resonance technique. This proposed colorimetric methodology has been applied to quantify the concentration of DO in real water samples. The result is consistent with the value obtained while adopting the traditional standard Winkler's methodology. A linear response of the absorbance of CdS QDs-GSH-MV system to the different concentrations of DO is obtained in the range of 0.75∼7.95 ppm, with a detection limit of 0.23 ppm in pH 7.5.
Introduction
Dissolved oxygen (DO) is an important parameter in various fields such as environment and biomedicine, which can be used as an essential indicator of water pollution, and is also an important support in blood analysis and metabolism monitoring in aerobic systems.1,2 The Winkler titration methodology, as the conventional way of the DO measurement, has been developed as the standard iodometric method for the determination (ISO 5813 Guide)3 and modified by Sakai et al.,4,5 Subsequently, an off-line Winkler's method coupled with flow injection analysis and an automatic flow system with in-line Winkler's procedure for the direct determination of DO in environmental waters was described.With the rapid development in the new environmentally-friendly methodologies including chemical,6 electrochemical7 or electron paramagnetic resonance (EPR)8 approaches, a great number of achievements have been made. The chemical method, in particular, the concentration of DO in water, was determined by the oxidation of manganese(II) to manganese(III) in alkaline medium and indicated by the formation of red manganese(III) ethylenediaminetetra-acetic acid complex in pH 4.0 buffer solution. Previous9,10 studies showed that electrogenerated superoxide ions managed to be utilized in oxygen determination in analytical progress. With the high sensitivity and operational simplicity, the electrochemical method might be preferred.11–14 Clark electrode is the most widely applied in quantifying the amount of oxygen in amperometric mode, with a problem that the electrode is rather sensitive to convection changes.15 However, since the electrodes could be easily poisoned, it is not proper to operate for a prolonged time in hostile or corrosive environments by means of the electrochemical methods. A recent literature reported on the attempted use a nanocrystal construction for oxygen sensing based on a fluorescence resonance energy transfer (FRET) signal transduction mechanism.16 The measurement of oxygen in vivo using EPR17 techniques based on the effect of oxygen on EPR spectra provides a sensitive and accurate means to measure oxygen quantitatively. However, it is time consuming and not conventional for real time and in situ measurement. A simple fluorescent probe has been developed recently for the determination of DO.18 Activated by iron(II) ions, oxygen could oxidize nonfluorescent coumarin to fluorescent 7-hydroxycoumarin. In addition, optical sensors19–23 entrapped in gel membranes were also developed for the determination of DO according to the fluorescence quenching induced by oxygen.24,25 But the gel membranes have poor mechanical property and the poor reproducibility of the membrane preparation. A photoelectrochemical sensor for DO based on poly(3,4-ethyllenedioxythiophene)/iron oxalate hybrid electrode was designed, depending on the photocurrent response to the oxygen concentration in the electrolyte.15
In order to meet the existing requirement for improving the determination of the low concentration of DO in real water samples, colorimetric sensing system turns out to be a good alternative. The concentration of oxygen could be determined according to the presence of a colorless compound that was produced by the reaction between colored reducing agents and oxygen.26–28 Quantum dots can make reversible changes between colors upon optical stimulation.29–33 In most cases, a colorless state was transformed to a colored one in response to light irradiation. The pronounced absorbance changes in the visible region of the absorption spectrum are responsible for the coloration and decoloration processes. As a result, quantum dots can be exploited efficiently to mediate the interplay of optical signals on the basis of absorptive and dispersive effects.34–37 Methyl viologen is widely used as an electron-transfer mediator in the studies of biological, chemical, and photochemical redox reactions.38 In the presence of electron donor GSH, the light irradiation of CdS QDs could induce MV2+ dication to be reduced to the corresponding deep blue radical cation.39,40 MV2+ dication's absorption peak around 260 nm would disappear while the reduced MV+˙ characterized by two absorption peaks at 396 and 606 nm arise.41
In our research, we demonstrated a new colorimetric method for simple and sensitive detection of DO based on the photochromic system of CdS QDs-GSH-MV. The principle of the method lies in observing the color change from deep blue to light yellow that occurs when a solution of MV+˙ is completely oxidized by oxygen in the test sample. The MV+˙ has one of the lowest redox potentials among all reported reversible organic redox systems (Eo′ = −0.44 V at pH 7.0) and reacts with oxygen rapidly.42 Experiments in UV-Vis absorbance spectra suggest that it is feasible for the determination of DO in real samples, and the obtained results are consistent with those obtained from the standard Winkler's method. In addition to its advantages of both the colorimetric approach and photochromic method, this approach could be applied in real time and in situ with a low detection limit, overcoming many problems related to the existing oxygen demand techniques. Therefore, this method could become a potential alternative for the detection of DO.
Experimental
Materials
All reagents were analytical grade. L-glutathione (GSH, 98.0∼101.0%), cadmium chloride hemi(-pentahydrate) (CdCl2, 99%), and thiourea (99%) (Sigma Chemical Co.) were used to prepare CdS QDs. Sodium phosphate monobasic, sodium phosphate dibasic and sodium hydroxide (Alfa Aesar China Ltd.) were employed to prepare the buffer solution (pH 6.0, 6.5, 7.0, 7.5, 8.0, 9.0, 10.0). Methyl viologen (1,1′-dimethyl-4,4′-bipyridinium dication or paraquat), sodium thiosulfate (Na2S2O3), sodium iodate (KIO3), and manganous chloride tetrahydrate (MnCl2·4H2O) were purchased from Aldrich (Milwaukee, WI) and were used without purification. Ultrapure water (18.2 MΩ) obtained from a WaterPro water purification system (Labconco Corpration, Kansas City, MO, USA) was used throughout. All other reagents and materials involved were also obtained commercially from Sinopharm Chemical Reagent Co. Ltd. (SCRC) and used as received without further purification.Synthesis of water soluble CdS QDs
0.2 mmol of CdCl2 solution and 0.32 mmol of GSH solution were mixed in 50 ml of distilled water. The pH of the solution was adjusted to 10.0 by 0.5 M NaOH. Then 0.38 mmol of thiourea was added to the solution with continuous vigorous stirring under N2 atmosphere. Subsequently, the reaction mixture was heated to 90 °C and remained for 6 h. The initial colorless solution turned bright yellow suggesting the formation of CdS QDs. Mean particle size and size distribution were monitored by the optical properties of CdS QDs through the temporal evolution of UV-visible and photoluminescence (PL) spectra. Finally, CdS QDs were precipitated by addition of acetone and then centrifuged after the solution was cooled to room temperature. The CdS QDs powder was obtained after drying in N2 atmosphere.Apparatus
UV-Vis absorption spectrum was obtained with Ocean Optics USB2000+ (190∼1700 nm) UV-Vis Miniature Fiber Optic Spectrometer using a high-performance 2048-element linear CCD-array detector. The EPR spectrum was performed in the X-band region with a Brucker EMX-8/2.7 spectrometer (UCL, Brussels, Belgium) at room temperature. EPR scan parameters were used as the following: microwave frequency 9.83 GHz, modulated frequency 100 KHz, modulated amplitude 0.50 G, time constant 163.840 s, conversion time 40.960 s; The pH value of a solution was measured by a PHS-3C (Switzerland Mettler Toledo Delta 320 pH meter); PL emission and excitation spectra were obtained on a Shimadzu Cary Eclipse (Varian) fluorometer; A Xenon lamp light source (PLS-SXE300, Bofeilai tech Co. Ltd. Beijing) was used. The cell was irradiated from a Xenon lamp operated at 300 W, through a home-made light chopper. The shutter chopping frequency was controlled manually.UV-Vis absorption spectroscopy
500 μl of 50 mM GSH, 50 μl of 50 mM MV2+, 150 μl of 2.98 μM CdS QDs, and 1 ml of PBS (pH 7.5) were added to a sealed cuvette (4 ml) and deaerated for 5 min. The mixture was continuously illuminated upon UV-Vis light while in a horizontally perpendicular direction to the light path, the absorbance intensity was recorded every 15 s for a total time of 6 min until the solution was changed completely into the deep blue. Then, 1 ml of different pure water samples was gently injected into the above-mentioned solution and the change of absorbance intensity was recorded according to the different oxygen concentrations. For determination of DO, 1 ml of different water samples was added carefully into the bottom of pH 7.5 PBS of CdS QDs-GSH-MV sealed blue solution with syringe, respectively. The change of UV-Vis spectrum of sealed CdS QDs-GSH-MV system depending on the concentration of DO in water samples were recorded within 6 min.Electron paramagnetic resonance (EPR) measurement
The solution containing 10 mM GSH, 10 mM MV2+, 1.78 μM CdS QDs, 0.2 M PBS was illuminated for 6 min by light irradiation until the blue color appeared and then the solution was sealed in an EPR quartz tube for radical measurement.Water samples
Four river water samples were obtained from campus and Huangpu River in Shanghai, China. Two sea water samples were also obtained from East China Sea, China. The sample vessel was submerged to a 30 cm depth to collect the surface water. The vessel was sealed after removing bubbles. The sampling bottles with narrow mouths designed to accommodate ground-glass stoppers were slightly oversized. The end of the glass stoppers were ground to a semiconical shape to prevent trapping of air bubbles when replacing stoppers. Two tap water samples were supplied by the water supply system on campus. Two pure water samples were obtained from a Milli-Q (Millipore) system. Two mineral water samples were supplied by two different mineral water production companies. Rigorous pretreatment was preformed prior to the analysis for all the samples.Preparation of different oxygen concentration
All oxygen must be removed from the sampling system with pure nitrogen. The bottles of 100 ml nominal volume were individually calibrated before the measurement was started. With a tube extending to the bottom of bottles, the sample overflows through an outlet tube into the bottle intake tube. In the measurement, the 10 ml glass syringe and the needle were rinsed with pure nitrogen several times. The prepared syringes with standard scale were used to aspirate a specified volume of high purity oxygen and sealed tightly. The oxygen gently injected specified volume oxygen into the pure water and then the water samples were purged with pure nitrogen for several minutes to make the various oxygen containing solutions. The actual oxygen concentration in water samples were carefully calibrated by the standard Winkler method.Determination of DO with standard Winkler method
The standard Winkler method was used to measure the DO content.43 The oxygen concentration obtained was taken as the standard value of DO in the sample.Results and discussion
Photochemistry of the CdS QDs-GSH-MV system
We have evaluated the spectroscopic properties of CdS QDs in the presence of MV2+ as electron acceptor and GSH as electron donor. The UV-Vis absorption spectrum of CdS QDs have a sharp peak centered around 360 nm (Fig. 1a), indicating that the distribution of particle size is relatively narrow and the size of particle is almost homogeneous. Fig. 1b depicts the emission spectra of CdS QDs with a pronounced peak at 500 nm. The fluorescence intensity decreases substantially with the increasing concentration of MV2+. When the concentration of MV2+ is up to 8.98 mM, the fluorescence is completely quenched (Fig. 1c). The effective process of luminescence quenching could be explained by the electron transfer from excited CdS QDs to MV2+.44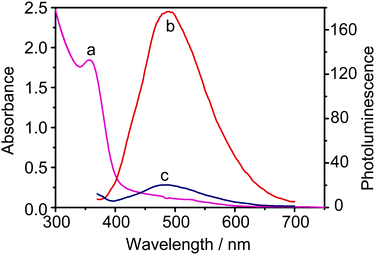 | ||
| Fig. 1 The absorption spectrum (a) of CdS QDs (1.78 μM) in 0.2 M PBS solution and emission spectra (λex = 360 nm) of the same solution recorded before (b) and after (c) the addition of MV2+ (8.98 mM). | ||
Fig. 2a records the absorption spectrum of CdS QDs-GSH-MV system without illumination in the degassed solution. After continuous irradiation for 45 s, two new absorption bands (Fig. 2b) appear at λ = 396 nm and 606 nm, which indicates the formation of radical cation of methyl viologen (MV+˙), and simultaneously the color changed from light yellow to light blue. After irradiation for about 300 s, two sharp peaks at about 396 and 606 nm could be observed (Fig. 2c). This phenomenon confirms charge transfer dominating the formation of deep blue free radical cation (MV+˙). The photoinduced coloration of CdS QDs-GSH-MV system is also evident from the increase of absorption at 606 nm with the increasing of irradiation time (Fig. 2d). When continuous irradiation was applied for a prolonged time, no absorption change was observed, which indicates that MV2+ in the solution is reduced to MV+˙ in maximum capacity. Moreover, the presence of radical cation is further verified and the stability of the system is also confirmed by EPR technique in subsequent experiment. The MV2+ is a suitable electron acceptor as it could easily accept one electron to produce a long-life free radical ion.45,46 In fact, the excited CdS QDs could efficiently collect and transfer electrons from donors to organic acceptors upon excitation. Scheme 1 shows the presumable schematic diagram (a, left) and the essence of chemical reaction of CdS QDs-GSH-MV system (b, left).
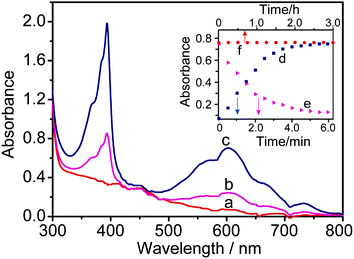 | ||
| Fig. 2 The absorption spectra of a system of CdS QDs (0.59 μM)-MV (9.62 mM)-GSH (1.54 mM) in 0.2 M PBS (pH = 7.5) recorded before (a) and after UV light irradiation for 45 s (b), 360 s (c); The evolution of the absorbance at 606 nm during continuous irradiation from 0∼360 s (d) and the subsequent exposure to oxygen (e). Time course of the absorbance spectra of CdS QDs-GSH-MV+˙ system without light irradiation in 0.2 M PBS (pH 7.5) (f). | ||
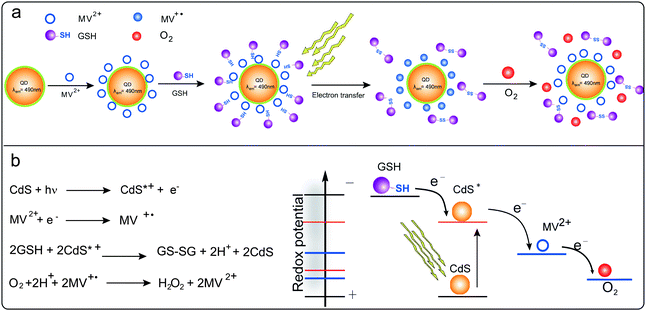 | ||
| Scheme 1 Schematic of the interaction of CdS QDs with MV2+ and GSH in the prepared CdS QDs-GSH-MV system (a); and the chemical reaction of overall CdS QDs-GSH-MV system (b, left) and the schematic of redox potentials of the photoelectrochemical system (b, right). The first electron transfer from photoactivated CdS QDs to MV2+ and the second step of electron transfer from sacrificial donor GSH to CdS QDs to fill the photogenerated hole. | ||
Sensitivity of DO to MV+˙
The photogenerated and colored MV+˙ could be changed back to the colorless MV2+ simply by exposing the sample to oxygen for a prolonged time. It is found that the gradual diffusion of oxygen in the samples induced a concomitant decrease in absorption at 606 nm (Fig. 2e). Meanwhile, color changed from deep blue to light yellow slowly (Scheme 1a, right). It is proposed that the deep-blue radicals (MV+˙) reacts with oxygen to produce the oxidized methyl viologen (MV2+). Observed color changes could be explained by the electron transfer from MV+˙ to oxygen. The potential mechanism has been proposed for the oxidation of MV+˙ (Scheme 1b, left) and the schematic of the redox potentials within this photoelectrochemical system is shown in Scheme 1b right. In this system, the hydrogen peroxide is generated from oxygen via the superoxide ion which is generated by the rapid reaction of MV+˙ with oxygen47 and superoxide ion reacts with H+ to give the HO2˙ radical under the experimental conditions employed.48 As is described in the previous section, the photoinduced rapid formation of the MV+˙ proceeds in deaerated aqueous solution with illumination. When oxygen gas was flushed into the mixture, the color of the MV+˙ rapidly faded (see the ESI†) and the simultaneous formation of hydrogen peroxide took place in the bulk solution. This suggests that the generation of hydrogen peroxide by the reaction of the MV+˙ with DO in the system.Based on the above discussion, the variety of absorbance intensity with the increasing oxygen content could offer a rapid method for the determination of DO in aqueous solutions.
Photostability of CdS QDs-GSH-MV system
The photosensitized reaction results in the formation of the MV+˙ under continuous irradiation. As shown in Fig. 2f, for CdS QDs-GSH-MV+˙ system, the absorbance spectra of resulting MV+˙ remain unchanged after three-hour storage without illumination, indicating that the free radical is very stable, which was produced through electron transfer mediated by CdS QDs under light illumination. The potential process of reaction could be explained by the electron transfer from the conduction band of the CdS QDs to MV2+ upon excitation of the transition involving the lowest electron state in CdS QDs.49,50 Thus, the observed little change of absorption spectra for CdS QDs-GSH-MV+˙ system further could confirm that the CdS QDs have better photostability and strong binding capability with MV2+ adsorbed tightly on the surface of nanoparticles.EPR measurement for MV+˙
EPR spectroscopy was employed in order to further validate the existence of the MV+˙. It is a highly specific and sensitive technique for direct detection of free radicals. In our research, the one-electron reduction of the MV2+ to form free radicals in aqueous solution is examined. As shown in Fig. 3, the recorded EPR spectrum (a) is very similar to the simulated one (b). For CdS QDs-GSH-MV system, after deoxygenization by high-purity nitrogen, under illumination in situ, the solution gradually changed into deep blue. Simultaneously, greater signal and clear EPR spectrum of MV+˙ could be observed, which are distinguished obviously (Fig. 3). Knowledge of the g-factor could give information about a paramagnetic center's electronic structure. This is especially significant for chemical systems. g value is determined in an EPR experiment by measuring the field and the frequency at which resonance occurs. The g value of MV+˙ is very close to that of the free electron, 2.0023.51 These results indicate that MV2+ is reduced into MV+˙ via the photoinduced electron transfer. However, there is no signal of radicals recorded in the system without CdS QDs or any electron donors before or after light illumination. Evidently, the electron obtained by MV2+ comes only from the donor of GSH. Whereas, it could be seen that in the presence of oxygen, MV+˙ can immediately transfer the electron to oxygen to form hydrogen peroxide and MV2+ dication, inducing the EPR signal of MV+˙ attenuated or disappeared inevitably.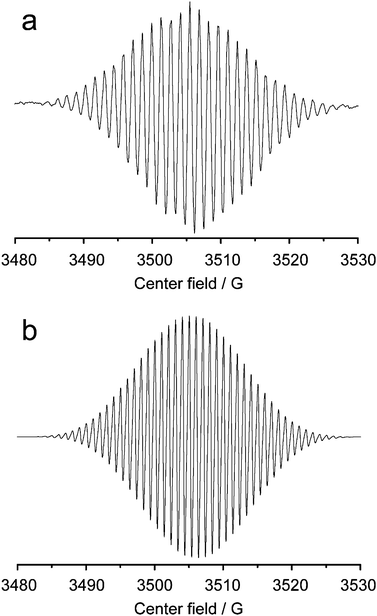 | ||
| Fig. 3 Electron paramagnetic resonance spectra of MV+˙, (a) recorded (b) simulated. | ||
From the above results, we could draw the conclusion that the electron transfer is induced by the excited quantum dots with the electron-hole pairs. The conduction-band holes donate electrons to MV2+ dication and then the valence-band holes oxidize GSH. The overall result is the photoinduced transfer of one electron from the sacrificial electron donor to the MV2+ dication acceptor with the formation of the colored radical cation. It is observed that the absorption spectrum of this system is dependent on the concentration of DO. Thus, the method for measuring oxygen content in real samples is designed based on the sensitive change of absorption with the concentration of DO.
UV-Vis absorbance spectra of CdS QDs-GSH-MV system in the presence of DO
In order to accurately determine the DO in real samples , the UV-Vis absorbance spectra of MV2+ and MV+˙ with the different concentration of DO were recorded. Firstly, the system was deaerated for 5 min. After the solution was irradiated for 6 min and MV2+ had been completely reduced into the stable deep blue MV+˙ radical state. When series of water samples with different concentrations of DO were added into the CdS QDs-GSH-MV+˙ solution, the change of absorption intensity was recorded immediately. The absorption spectra changes with the addition of different concentrations of oxygen are shown in Fig. 4. Increasing oxygen content induced a gradual absorption decrease of MV+˙ at 606 nm and the color changed apparently from deep blue to light yellow. With the increasing concentration of oxygen, additional absorption peaks appear in the visible region at about 260 nm while the absorption peaks at approximately 396 and 606 nm disappear, which also imply that the reoxidized of MV+˙ induced by the addition of oxygen. It is found that DO content in solution has a good linear relationship with the absorption intensity of MV+˙ at 606 nm. Therefore, this method is established successfully to determine the DO in real samples according to the change of absorption spectra with different content of DO.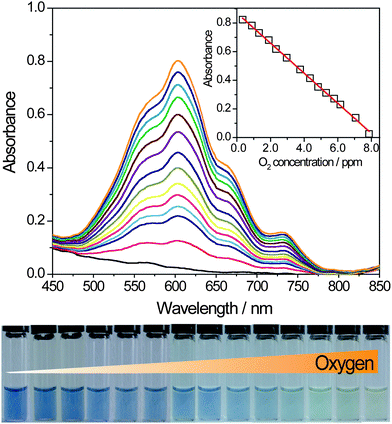 | ||
| Fig. 4 The UV-Vis absorbance spectra of CdS QDs-GSH-MV system with the different content of DO in the PBS (pH = 7.5) solution, CMV2+ = 1 mM, CCdSQDs = 0.59 μM, CGSH = 10 mM, from top to bottom, the added concentration of oxygen from 0.75∼7.95 ppm (from left to right in the photo). | ||
Based on the sensitivity of oxygen to the absorption spectra of MV+˙, we employed UV-vis spectrometry to quantitatively determine the DO in water samples. It could be seen that the absorption intensity of the system decreased noticeably with the increasing concentration of DO. The reduced intensity of absorption at 606 nm was in proportion to the concentration of DO in the range of 0.75∼7.95 ppm, the linear regression equation was Iabs = −0.107C (ppm) + 0.890 with a correlation coefficient (R) of 0.9987. The detection limit for oxygen was determined to be 0.23 ppm. The relative standard deviation (RSD) is obtained from 5 repeated determinations.
Effect of pH on the rate of photoreduction of MV2+
The effect of pH on the photoreduction of MV2+ was carried out with a solution of 0.59 μM CdS QDs, 10 mM GSH, and 1 mM MV2+ in various PBS buffer. The variation in the rate of photoreduction of MV2+ with pH is given in Fig. 5, at a constant lumination. It could be seen from these data that the maximum rate of MV+˙ formation takes place at pH 7.5. It might be explained that the CdS QDs is more stable at this pH than others under the experimental conditions of our research.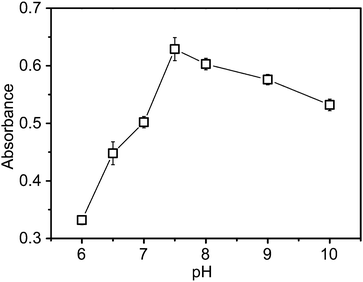 | ||
| Fig. 5 Effect of pH on the rate of photoreduction of MV2+ with GSH as the electron donor, CMV2+ = 1 mM, CCdSQDs = 2.98 μM, CGSH = 10 mM. Each error bar indicates the standard deviation associated with five different measurements. | ||
These particles will be negatively charged and could form stable complexes with MV2+, which make the photoelectrochemical reaction proceed easily.
Effect of MV2+ concentration
The effect of MV2+ concentration is investigated in the range from 0.1 to 20 mM. The absorption intensity of the system increases gradually with addition of higher concentrations of MV2+. The optimum amount of MV2+ here is found to be 1 mM. We would not adopt a larger concentration for the poor linearity between the absorbance and the quantity of MV+˙.DO sample analysis
The DO contents obtained by both methods in twelve practical samples arranged according to sampling stations are presented in Table 1. As illustrated in Table 1, the relative error is less than 5% between the proposed new method and the standard Winkler's method. It means that there is no obvious difference between these two methods. The F-test with 95% confidence level was carried out to contrast the results obtained by the two methods.18 The results of the randomization F-test indicate that all samples are not significant for their F-values, which are lower than the tabulated critical value (F (4, 4) = 6.39) at the 95% confidence level, indicating that the two methods agreed very well. This method, therefore, provides a good alternative for the determination of DO for real samples.| Samples | Colorimetric method (ppm) | Winkler's methoda (ppm) | Relative Standard Error (%) | F values in F-testb |
|---|---|---|---|---|
| a Take the DO values by Winkler's method as standards. b For comparison, F(4, 4) = 6.39. | ||||
| Sea water I | 7.14 ± 0.18 | 7.65 ± 0.03 | −1.14 | 2.27 |
| Sea water II | 7.29 ± 0.14 | 7.43 ± 0.05 | 0.64 | 3.46 |
| Huangpu river water I | 6.26 ± 0.11 | 6.37 ± 0.10 | −1.17 | 2.89 |
| Huangpu river water II | 6.48 ± 0.19 | 6.83 ± 0.07 | 1.70 | 4.23 |
| Campus river water I | 7.56 ± 0.13 | 7.38 ± 0.12 | 2.44 | 3.56 |
| Campus river water II | 7.77 ± 0.17 | 7.86 ± 0.09 | −1.15 | 2.67 |
| Tap water I | 5.21 ± 0.12 | 5.03 ± 0.04 | 3.58 | 4.02 |
| Tap water II | 5.26 ± 0.16 | 5.34 ± 0.08 | −1.50 | 3.91 |
| Pure water I | 5.07 ± 0.15 | 5.17 ± 0.11 | −1.93 | 2.73 |
| Pure water II | 4.99 ± 0.19 | 4.87 ± 0.02 | 2.46 | 4.12 |
| Mineral water I | 4.89 ± 0.17 | 4.86 ± 0.09 | 0.63 | 3.48 |
| Mineral water II | 4.49 ± 0.13 | 4.64 ± 0.06 | −3.23 | 4.61 |
Interferences in the determination of DO
A systematic investigation of the possible interference has been carried out as well. Fe3+, Cu2+, and Ca2+ were tested for possible interferences and the results are given in Table 2. Fe3+ gives positive errors by the Winkler's method due to oxidation of iodide by Fe3+. Fe3+ also gives positive errors by the Colorimetric method because MV+˙ could be oxidized into its original state MV2+ by Fe3+.52 Thus, in order to make the determination results more accurately, the content of Fe3+ in real samples should be sheltered. Results show that the determination of real samples with Winkler's method and colorimetric method is not affected by the co-existing ions of Ca2+ and Mg2+. For Mn2+, Co2+ and Ni2+, these transition metal ions could directly quench the fluorescence of CdS QDs and might prevent the production of electron transfer. However, the average content of these co-existing metal ions do not exceed the tolerant concentrations in the tap water.| Cation | C (ppm) | Winkler's method (ppm) | Colorimetric method (ppm) |
|---|---|---|---|
| Fe3+ | 0.3 | 7.44 | 7.52 |
| Cu2+ | 1.0 | 6.78 | 6.45 |
| Zn2+ | 1.0 | 6.23 | 6.56 |
| Ca2+ | 250 | 7.31 | 7.25 |
| Mg2+ | 250 | 7.28 | 7.20 |
| Co2+ | 0.1 | 6.34 | 6.45 |
| Ni2+ | 0.02 | 6.19 | 6.21 |
| Mn2+ | 0.2 | 5.88 | 5.71 |
Conclusions
In summary, a colorimetric analytical principle for the detection of DO has been proposed and experimentally validated. It is successfully applied to determinate the DO concentrations in the real samples based on photochemical generation of MV+˙ in CdS QDs-GSH-MV system at room temperature. CdS QDs can efficiently mediate the transfer of electrons from sacrificial donor GSH to MV2+ upon light irradiation. The existence of deep blue MV+˙ is verified by EPR in the degassed solution. MV+˙ could react rapidly with oxygen to produce the original oxidized MV2+. The absorbance intensity of MV+˙ at 606 nm respond well with the concentration of DO. The results of DO concentrations obtained from this method are in good agreement with those from Winkler's method. Under the optimized conditions, a better linear correlation is established between the absorption intensity and the concentration of DO in the range of 0.75∼7.95 ppm with a detect limit of 0.23 ppm. It is also rapid, and easy to be automated. Thus, high sensitivity and low detection make the proposed method feasible for the rapid analysis of DO.Acknowledgements
This work was supported by National High Technology Research and Development Program of China (Grant No. 2008AA06A406) and Shanghai Pujiang Program Grant of China (Grant No. 08PJ1403100 and 09PJ1403300).Notes and references
- L. K. Ju and P. B. Nallagatla, Water Res., 2003, 37, 188 CrossRef CAS.
- J. S. Ye, Y. Wen, W. D. Zhang, H. F. Cui, L. M. Gan, G. Q. Xu and F. S. Sheu, J. Electroanal. Chem., 2004, 562, 241 CrossRef CAS.
- International Standard Orgnisation, Water Quality–Determination of Dissolved Oxygen- Iodometric Method, ISO 5813, ISO, Geneva, 1983 Search PubMed.
- T. Sakai, H. Takio, N. Teshima and H. Nishikawa, Anal. Chim. Acta, 2001, 438, 117 CrossRef CAS.
- T. Sakai, S. Piao, N. Teshima, T. Kuroishi and K. Grudpan, Talanta, 2004, 63, 893 CAS.
- S. A. Rahim and S. H. Mohamed, Talanta, 1978, 25, 519 CrossRef CAS.
- G. Ilangovan, H. Q. Li, J. L. Zweier and P. Kuppusamy, J. Phys. Chem. B, 2001, 105, 5323 CrossRef CAS.
- Joseph A. Hofmann, Jr. and F. B. David, Inorg. Chem., 1984, 23, 1177 CrossRef.
- J. F. Coetzee and I. M. Kolthoff, J. Am. Chem. Soc., 1957, 79, 6110 CrossRef.
- J. E. A. Toni, J. Electrochem. Soc., 1969, 116, 212 CrossRef CAS.
- J. B. Zimmerman and R. M. Wightman, Anal. Chem., 1991, 63, 24 CrossRef CAS.
- D. Obendorf and M. Wilhelm, Anal. Chem., 2003, 75, 1374 CrossRef CAS.
- R. D. S. Luz, F. S. Damos, A. A. Tanaka and L. T. Kubota, Sens. Actuators, B, 2006, 114, 1019 CrossRef.
- R. N. Gillanders, M. C. Tedford, P. J. Crilly and R. T. Bailey, Anal. Chim. Acta, 2005, 545, 189 CrossRef CAS.
- G. Bencsik, Z. Lukács and C. Visy, Analyst, 2010, 135, 375 RSC.
- E. J. McLaurin, A. B. Greytak, M. G. Bawendi and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 12994 CrossRef CAS.
- H. M. Swartzy and R. B. Clarksonz, Phys. Med. Biol., 1998, 43, 1957 CrossRef CAS.
- W. Luo, M. E. Abbas, L. H. Zhu, W. Y. Zhou, K. J. Li, H. Q. Tang, S. S. Liu and W. Y. Li, Anal. Chim. Acta, 2009, 640, 63 CrossRef CAS.
- C. M. Mcdonagh, B. D. Maccraith and A. C. Mcavoy, Anal. Chem., 1998, 70, 45 CrossRef CAS.
- R. T. Bailey, F. R. Cruickshank, G. Deans, R. N. Gillanders and M. C. Tedford, Anal. Chim. Acta, 2003, 487, 101 CrossRef CAS.
- P. A. S. Forge, P. Caldas, C. C. Rosa, A. G. Oliva and J. L. Santos, Sens. Actuators, B, 2004, 103, 290 CrossRef.
- R. M. Bukowski, R. Ciriminna, M. Pagliaro and F. V. Bright, Anal. Chem., 2005, 77, 2670 CrossRef CAS.
- S. Sax, E. Fisslthaler, S. Kappaun, C. Konrad, K. Waich, T. Mayr, C. Slugovc, I. Klimant and E. J. W. List, Adv. Mater., 2009, 21, 3483 CrossRef CAS.
- M. M. F. Choi and D. Xiao, Anal. Chim. Acta, 1999, 387, 197 CrossRef CAS.
- L. Guo, Q. Ni, J. Li, L. Zhang, X. Lin, Z. Xie and G. Chen, Talanta, 2008, 74, 1032 CrossRef CAS.
- R. C. Evans and P. Douglas, ACS Appl. Mater. Interfaces, 2009, 1, 1023 Search PubMed.
- R. C. Evans and P. Douglas, Anal. Chem., 2006, 78, 5645 CrossRef CAS.
- S. K. Lee, M. Sheridan and A. Mills, Chem. Mater., 2005, 17, 2744 CrossRef CAS.
- S. M. Emin, N. Sogoshi, S. Nakabayashi, T. Fujihara and C. D. Dushkin, J. Phys. Chem. C, 2009, 113, 3998 CrossRef CAS.
- Y. Kishimoto and J. Abe, J. Am. Chem. Soc., 2009, 131, 4227 CrossRef CAS.
- I. Yildiz and F. M. Raymo, J. Mater. Chem., 2006, 16, 1118 RSC.
- K. D. Belfield, Y. Liu, R. A. Negres, M. G. Fan, G. L. Pan, D. J. Hagan and F. E. Hernandez, Chem. Mater., 2002, 14, 3663 CrossRef CAS.
- J. Andersson, S. M. Li, P. Lincoln and J. Andréasson, J. Am. Chem. Soc., 2008, 130, 11836 CrossRef CAS.
- F. M. Raymo and M. Tomasulo, J. Phys. Chem. A, 2005, 109, 7343 CrossRef CAS.
- S. Silvi, A. Arduini, A. Pochini, A. Secchi, M. Tomasulo, F. M. Raymo, M. Baroncini and A. Credi, J. Am. Chem. Soc., 2007, 129, 13378 CrossRef CAS.
- E. Deniz, M. Tomasulo, S. Sortino and F. M. Raymo, J. Phys. Chem. C, 2009, 113, 8491 CrossRef CAS.
- F. M. Raymo and M. Tomasulo, J. Phys. Chem. A, 2005, 109, 7343 CrossRef CAS.
- T. W. Ebbesen, L. E. Manring and K. S. Peters, J. Am. Chem. Soc., 1984, 106, 7400 CrossRef CAS.
- H. Kamogawa and O. Toshihiko, Chem. Mater., 1991, 3, 1020 CrossRef.
- T. Kuwabara, H. Nakajima, M. Nanasawa and A. Ueno, Anal. Chem., 1999, 71, 2844 CrossRef CAS.
- T. Watanabe and K. Honda, J. Phys. Chem., 1982, 86, 2617 CrossRef CAS.
- T. Pérez-Ruiz, C. Martinez Lozano and V. Tomás, Mikrochim. Acta, 1985, 86, 367 CrossRef.
- J. H. Carpenter, Limnol. Oceanogr., 1965, 10, 141 CrossRef CAS.
- Y. Nosaka and M. A. Fox, J. Phys. Chem., 1988, 92, 1893 CrossRef CAS.
- A. Nakahara and J. H. Wang, J. Phys. Chem., 1963, 67, 496.
- L. Michaelis, Chem. Rev., 1935, 16, 243 CrossRef CAS.
- J. A. Farrington, M. Ebert, E. J. Land and K. Fletcher, Biochim. Biophys. Acta, Bioenerg., 1973, 314, 372 CrossRef CAS.
- H. Hormann, C. Neubauer, K. Asada and U. Schreiber, Photosynth. Res., 1993, 37, 69 CrossRef CAS.
- L. I. Halaoui, J. Electrochem. Soc., 2003, 150, E455 CrossRef CAS.
- H. B. Yildiz, R. Tel-vered and I. Willner, Angew. Chem., Int. Ed., 2008, 47, 6629 CrossRef.
- C. S. Johnson Jr. and K. S. Gutowsky, J. Chem. Phys., 1963, 39, 59.
- S. S. Hu, J. Electroanal. Chem., 1999, 463, 253 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Process of oxygen sensitive CdS QDs-GSH-MV system under light illumination. See DOI: 10.1039/c0ay00201a/ |
| This journal is © The Royal Society of Chemistry 2010 |