Development of an oxalate-affinity chromatographic column to isolate oxalate-binding proteins
Piyachat
Roop-ngam
ab and
Visith
Thongboonkerd
*ac
aMedical Proteomics Unit, Office for Research and Development, Faculty of Medicine, Siriraj Hospital, Mahidol University, 12th Floor Adulyadejvikrom Building, 2 Prannok Road, Bangkoknoi, Bangkok, 10700, Thailand. E-mail: thongboonkerd@dr.com; vthongbo@yahoo.com; Fax: +66-2-4184793; Tel: +66-2-4184793
bDepartment of Immunology and Immunology Graduate Program, Faculty of Medicine, Siriraj Hospital, Mahidol University, Bangkok, Thailand
cCenter for Research in Complex Systems Sciences, Mahidol University, Bangkok, Thailand
First published on 9th July 2010
Abstract
Calcium oxalate (CaOx) is the major crystalline component in kidney stones. Many previous studies on stone modulation have focused mainly to calcium-binding proteins, whereas oxalate-binding proteins remain under-investigated due to a lack of a simple method to purify oxalate-binding proteins. Therefore, we have developed an affinity chromatographic column to isolate or purify oxalate-binding proteins. The oxalate-affinity column developed contained EAH-Sepharose 4B beads conjugated with oxalic acid by carbodiimide reaction using 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC) as an activator. The coupling efficacy of coupled beads (oxalic acid-EAH Sepharose 4B) was determined by a competitive ninhydrin assay to quantify the remaining amino group on EAH-Sepharose 4B. In addition, the oxalate-affinity column showed high specific binding to a known oxalate-binding protein (p62) but not to an irrelevant protein (carbonic anhydrase). In conclusion, we have established the oxalate-affinity chromatographic column, which showed the high efficiency to isolate oxalate-binding proteins. This novel chromatographic column will be very useful for future stone research, particularly in the area of stone modulation by oxalate-binding proteins.
Introduction
Oxalate (C2O42−) is a naturally occurring substance produced and found in plants, animals and humans. It is an end product of metabolic processes, such as breakdown of glyoxylic acid or ascorbic acid, and under normal physiological conditions, is primarily excreted by the kidney. Oxalate plays an important role in kidney stone formation as a counter ion of calcium, both of which form calcium oxalate (CaOx) crystal that is the most common crystalline component found in kidney stones.1 There is an increasing amount of evidence demonstrating that abnormality of oxalate metabolism plays a vital role in kidney stone disease.2 Excessive excretion of oxalate into renal tubular fluid leads to CaOx crystalluria and renal injury in animal model.3 Oxalate-induced apoptosis of renal tubular epithelial cells can cause renal damage and may involve in an initial step of the pathogenic mechanisms of CaOx kidney stone disease.4 Moreover, alteration in renal epithelial oxalate transport pathways, especially oxalate transporters or oxalate-binding molecules, may exert in oxalate retention inside renal tubular epithelial cells.5Interestingly, several reports have shown that oxalate-binding proteins exhibit crystal modulating activity.6 A number of oxalate-binding proteins have been isolated from kidney homogenate and stone matrix.6 Some of them have been successfully identified, e.g. histone-H1B.7 Two-thirds of proteins with oxalate-binding ability found in rat kidney and liver homogenates are localized in nucleus, whereas the other one-third are present in mitochondria.7 The binding between nuclear proteins and oxalate has been proposed to mediate through a basic amino acid residue; i.e. lysine. In addition, the oxalate-binding bond can be disrupted by a lysine modifier, 4,4′-diisothiocyanostilbene 2,2′-disulfonic acid (DIDS).7
Because of their roles in CaOx crystal modulation, oxalate-binding proteins should be investigated more extensively, and isolation as well as purification of oxalate-binding proteins derived from renal tubular epithelial cells are crucial. Previously, oxalate-binding proteins were labeled by 14C-oxalate, which was used to bind with them, and were then purified by isolation of radioisotopic protein fractions using Sephadex and/or diethylaminoethyl (DEAE) columns.8 The oxalate-binding assay was performed by counting a radioisotope signal of proteins-bound 14C-oxalate. Although this conventional assay is highly sensitive, it is sophisticated and has to deal with radioisotope (14C), which could be its drawback. Because a simple method for isolation of oxalate-binding proteins was not well established in the past, we have developed an easy-to-use method with simplified purification steps as follows.
Results and discussion
Affinity chromatography is well known as a simple technique to purify proteins depending on their interaction to chemical structure of an immobilized specific ligand coupled to a chromatographic matrix. In this study, affinity chromatography was developed to isolate oxalate-binding proteins. Commercially available EAH-Sepharose beads 4B (GE Healthcare) was selected as the chromatographic material, which provided a primary amine group readily conjugated with a diaminohexane spacer for efficient coupling with oxalate ligand. Several studies have employed EAH-Sepharose 4B for coupling with biomolecules containing carboxylic acid (e.g. abscisic acid)9 or cholic acid.10 However, oxalate-affinity chromatographic columns for isolation and purification of oxalate-binding proteins were not well established in the past. In the present study, we therefore developed an oxalate-affinity chromatographic column by modification of the established abscisic acid (ABA)-linked EAH-Sepharose 4B column described previously by Zhang et al.9The method to covalently link between carboxylic and amino groups was previously described by O'Carra and Barry.11 Unlike our present study, they employed cyanogens bromide-activated Sepharose 4B as the material to establish an oxamate-affinity chromatographic column. The conjugation between carboxylic and amino groups in the oxamate-affinity column was mediated by a hexamethylene diamine spacer arm.11 In the present study, we used EAH-Sepharose 4B beads, which contained 10-atom hexamethylene diamine spacer arms for facilitating the covalent link between carboxylic and amino groups. The coupling reaction was mediated by 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) for carboxylic activation (Fig. 1A).
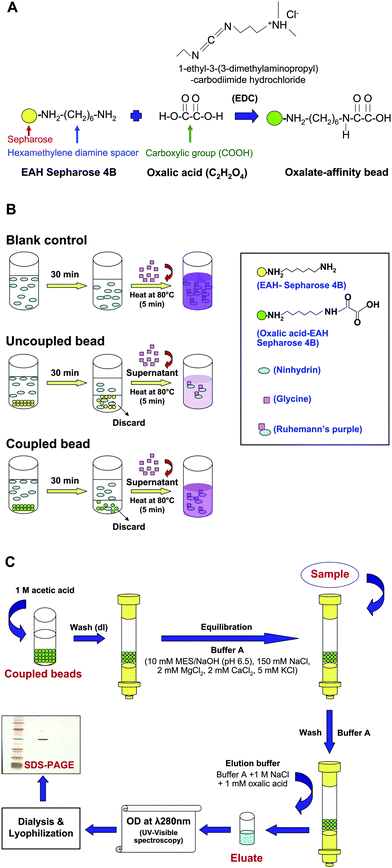 | ||
| Fig. 1 Schematic summary of methodology. (A) Coupling reaction of oxalic acid to EAH-Sepharose 4B bead. EAH-Sepharose 4B was conjugated with a carboxylic group (COOH) of oxalic acid (C2H2O4) through its hexamethylene diamine spacer (NH2(CH2)6NH2). The carboxylic activation was mediated by EDC (1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride). (B) Competitive ninhydrin assay. The oxalate-coupled or uncoupled beads were incubated with ninhydrin solution for 30 min and then discarded prior to a reaction with glycine, whereas ninhydrin solution without pretreatment with beads served as the blank control. While the amount of glycine was fixed, colorimetric measurement of the resulting Ruhemann's purple complex (at λ 570 nm) would reflect the degree of the reaction, which was determined by the remaining primary amine groups on the beads. The effective conjugation of oxalate on the beads would result in a minimal amount of free amine groups remaining on the bead surface to competitively bind to ninhydrin, thus providing the maximal amount of ninhydrin left for reaction with glycine to provide a strong Ruhemann's purple. In contrast, ineffective conjugation would result in a weak Ruhemann's purple. (C) Column preparation and isolation of oxalate-binding protein. Firstly, the remaining amine groups on oxalate-conjugated EAH-Sepharose 4B were blocked with 1M acetic acid and then washed extensively with deionized water (dI water). Thereafter, oxalic acid-EAH-Sepharose 4B beads were packed into the column. The oxalate-affinity chromatographic column was equilibrated with Buffer A before sample loading. After incubation and washing, the bound proteins were eluted and subsequently analyzed by UV-visible spectrophotometry (at λ 280 nm) and SDS-PAGE. | ||
Indeed, a similar strategy was previously used by Kim and colleagues12 to develop an oxalate-affinity chromatographic column. They performed oxalate-affinity chromatography after DEAE-Sephacel anion exchange chromatography and before hydroxyapatite chromatography to purify malate synthase from Acinetobacter calcoaceticus. However, as their focus was to purify an enzyme malate synthase, not to isolate oxalate-binding proteins, no known oxalate-binding proteins were examined as the positive control in their study. Moreover, they did not evaluate efficacy of the coupling reaction.
In our present study, the coupling efficacy was determined quantitatively by measuring the remaining primary amine groups on EAH-Sepharose 4B beads using a competitive ninhydrin assay (Fig. 1B). In principle, ninhydrin generally bound to free amino acid residues (i.e. glycine), give a reaction with Ruhemann's purple complex.13 On the other hand, ninhydrin could also bind to primary amine groups. Any residual free amine groups remaining on the EAH-Sepharose 4B beads following the oxalic acid coupling reaction would result in a reduced amount of ninhydrin available to react with glycine and this would be observed as a reduced intensity of the Ruhemann's purple. The effective conjugation of oxalate on the beads would result in minimal amount of free amine groups remaining on the bead surface to competitively bind to ninhydrin, thus providing the maximal amount of ninhydrin left for reaction with glycine to provide a strong Ruhemann's purple. In contrast, ineffective conjugation would result in a weak Ruhemann's purple. Fig. 2 clearly demonstrates that our oxalate-affinity EAH-Sepharose 4B beads had effective conjugation of oxalic acid with amine groups on the beads, and yielded significantly greater intensity of Ruhemann's purple (measured by UV-visible spectrophotometry at λ 570 nm), as compared to the unconjugated beads. Note that, during the initial phase of this study, our methods were optimized to obtained satisfactory coupling efficacy and specificity as shown herein.
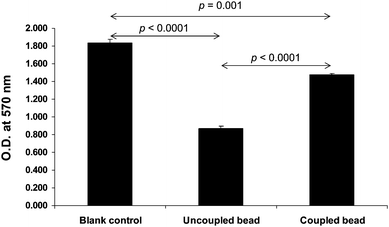 | ||
| Fig. 2 Determination of coupling efficacy by competitive ninhydrin assay. The competitive ninhydrin assay was performed as detailed in Fig. 1B. The data are reported as Mean ± SEM (n = 3 independent experiments). p values < 0.05 were considered statistically significant. | ||
The specificity of our oxalate-affinity beads for chromatographic protein isolation/purification was evaluated using p62 as the positive control and carbonic anhydrase as the negative control. Column and sample preparation was performed as detailed in “Materials and Methods” and analytical steps are summarized in Fig. 1C. In total, 10 eluate fractions (1 ml/fraction) were collected and their protein contents were measured by UV-visible spectrophotometry at λ 280 nm. The positive control, p62, which is a known oxalate-binding proteins,14 was detectable in the eluate fractions as shown by the measurable protein content (data not shown). In contrast, no protein content was found in the elution profile collected from the negative control carbonic anhydrase, indicating that there was no retention of carbonic anhydrase in the oxalate-affinity chromatographic column (data not shown). The results also suggest that the oxalate immobilized ligand retained its specific binding affinity with the target proteins after the coupling step. In addition, the binding between oxalate and the target proteins was reversible, or in other words, was able to be eluted by using a competitive ligand, which was a free form of oxalate in our study.
After salt removal, eluate fractions of the tested proteins were concentrated by lyophilization and then resolved in 12% SDS-PAGE under reducing conditions. The resolved proteins were visualized using silver stain. Consistent with the measurement of protein content, it was found that p62 (positive control) was present predominately at a molecular mass of ∼62 kDa from the eluate fractions obtained from oxalate-affinity chromatographic column (Fig. 3A), whereas no protein band of carbonic anhydrase (negative control) was found in its eluate fractions (Fig. 3B). The protein recovery yield of our oxalate-affinity chromatographic column to isolate p62 was approximately 12.5% (which was comparable to 11% recovery yield by combined three chromatographic steps to isolate malate synthase in the study by Kim and colleagues).12 Mass spectrometric analysis (using Q-TOF MS/MS) of the resolved protein bands in Fig. 3A derived from our oxalate-affinity chromatographic column identified p62 (or nucleoporin 62 kDa; NCBI ID: gi|24497603; MS/MS ions score: = 75; p < 0.05) in all bands visualized. These results from the oxalate-affinity chromatographic column revealed a unique selectivity in that it could bind specifically to oxalate-binding proteins, but not to irrelevant proteins.
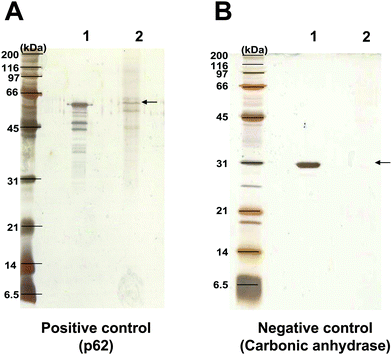 | ||
| Fig. 3 SDS-PAGE of the eluted proteins. p62 served as the positive control (A), whereas carbonic anhydrase served as the negative control (B). After loading of 10 μg each to the oxalate-affinity chromatographic column and incubation as summarized in Fig. 1C, the eluted proteins were resolved by 12% SDS-PAGE and visualized by silver staining. Lane 1, 2 μg of purified p62 or carbonic anhydrase. Lane 2, the eluate fractions of 10 μg of corresponding proteins derived from oxalate-affinity column chromatography. | ||
Conclusions
In summary, we have successfully established the oxalate-affinity chromatographic column for highly efficient and simplified isolation/purification of oxalate-binding proteins. This novel column will be very useful for future research on kidney stone disease, particularly in the study of CaOx crystal modulation by oxalate-binding proteins.Materials and methods
Preparation of oxalate-affinity beads
Oxalic acid (Sigma-Aldrich; Singapore) was coupled (through its carboxylic group) to EAH-Sepharose 4B beads (GE Healthcare; Uppsala, Sweden) containing 10-atom diaminohexane spacers, which had been readily conjugated with primary amine groups (7–11 μmol per 1 ml of drained gels). Fig. 1A summarizes the coupling reaction of oxalic acid to EAH-Sepharose 4B that was performed by dissolving 0.24 g of oxalic acid in 3.6 ml of 50% (w/v) dimethylformamide solution (Merck; Victoria, Australia) mixed with 3 ml of drained EAH-Sepharose 4B. To activate the carboxylic group of oxalic acid, 0.23 g of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) (Merck) was added to the oxalic acid/EAH-Sepharose 4B mixture and the pH was adjusted to 8.0 with 20 M NaOH. The mixture was then stirred gently at 4 °C overnight in the dark. After the coupling reaction, the oxalic acid-EAH-Sepharose 4B beads were washed with 50% (w/v) dimethylformamide (Merck) and then with 0.5 M NaCl in 0.1 M Tris-HCl buffer (pH 8.3), followed by 0.5 M NaCl in 0.1 M sodium acetate-acetic acid buffer (pH 4.0). Thereafter, the beads were extensively washed with deionized (dI) water (18.2 MΩ·cm water). The uncoupled EAH Sepharose 4B beads, which were incubated with 1 M acetic acid at 4 °C overnight and subsequently washed with dI water, served as the control.Determination of coupling efficacy
Coupling efficacy was determined quantitatively by measuring the remaining primary amine groups on EAH-Sepharose 4B beads using a competitive nindydrin assay (Fig. 1B). Briefly, 10 μl of 20 mM ninhydrin was mixed with 80 μl dI water. Then, 10 μl of oxalate-conjugated or unconjugated EAH-Sepharose 4B beads was added into the ninhydrin solution. After a 30-min incubation on a continuous rotator at room temperature (RT), the beads were discarded by a centrifugation at 313 g for 5 min and the supernatant was reacted with 150 μl of 20 mM glycine. The reaction was performed at 80 °C for 5 min and cooled down at RT for 10 min before measurement of the Ruhemann's purple complex by UV-visible spectrophotometry at λ 570 nm (Shimadzu; Kyoto, Japan). The blank control was processed as aforementioned, but without incubation with beads prior to reaction with glycine.Column preparation and isolation of oxalate-binding proteins
After determination of coupling efficacy, the remaining amine groups on oxalic acid-EAH Sepharose 4B were blocked by an overnight incubation with 1 M acetic acid and then extensively washed with dI water before being packed into a 1 × 10 cm, 8 ml of Glass Econo-Column® (Bio-Rad Laboratories; Hercules, CA). The column was equilibrated with 20 ml of Buffer A containing 10 mM 2-morpholinoethanesulfonic acid (MES) in NaOH (pH 6.5), 150 mM NaCl, 2 mM MgCl2, 2 mM CaCl2, and 5 mM KCl. After equilibration, 10 μg of individual proteins in Buffer A was loaded into the column and incubated overnight at 4 °C. p62 (Abcam; Cambridge, UK), was used as a positive control, whereas carbonic anhydrase (Sigma-Aldrich), which has no oxalate-binding property, was used as a negative control. Thereafter, the column was washed with 5 ml of Buffer A to remove unbound proteins. Oxalate-binding proteins were then eluted with 10 ml of elution buffer (Buffer A with 1 M NaCl and 1 mM oxalic acid) and fractionated into 10 fractions (1 ml/fraction). The eluted protein fractions were monitored for protein content at λ 280 nm by UV-visible spectrophotometer. The protocol for isolation of oxalate-binding proteins is summarized in Fig. 1C.SDS-PAGE separation of oxalate-binding proteins
The eluted protein fractions were pooled and desalted by dialysis overnight against dI water. The dialyzed protein fractions were concentrated by lyophilization, resuspended with 1X Laemmli buffer, and heated at 95 °C for 5 min. Proteins were then resolved with 12% SDS-PAGE at 150 V for approximately 2 h using SE260 mini-Vertical Electrophoresis Unit (GE Healthcare). The resolved protein bands were then visualized by silver stain.Protein identification by mass spectrometry
The resolved protein bands were excised and subjected to in-gel tryptic digestion as described in our previous study.15 The trypsinized samples were then analyzed by quadrupole time-of-flight (Q-TOF) tandem mass spectrometry (MS/MS). Briefly, the trypsinized samples were premixed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with the matrix solution containing 5 mg ml−1 α-cyano-4-hydroxycinnamic acid in 50% ACN, 0.1% (v/v) TFA and 2% (w/v) ammonium citrate, and deposited onto the 96-well MALDI (matrix-assisted laser desorption/ionization) target plate. The samples were analyzed by Q-TOF Maxis™ mass spectrometer (Bruker Daltonik GmbH; Billerica, MA). The obtained MS/MS ions data were analyzed using MASCOT search engine (http://www.matrixscience.com) to query to the NCBI mammalian protein database, assuming that peptides were monoisotopic. Fixed modification was carbamidomethylation at cysteine residues, whereas variable modification was oxidation at methionine residues. Only one missed trypsin cleavage was allowed. Peptide mass tolerance was 1.2 Da, whereas fragmented mass tolerance was 0.6 Da. MS/MS ions scores >47 were considered statistically significant (significant hits). More details of Q-TOF MS/MS analysis are described in our previous study.15
:1 with the matrix solution containing 5 mg ml−1 α-cyano-4-hydroxycinnamic acid in 50% ACN, 0.1% (v/v) TFA and 2% (w/v) ammonium citrate, and deposited onto the 96-well MALDI (matrix-assisted laser desorption/ionization) target plate. The samples were analyzed by Q-TOF Maxis™ mass spectrometer (Bruker Daltonik GmbH; Billerica, MA). The obtained MS/MS ions data were analyzed using MASCOT search engine (http://www.matrixscience.com) to query to the NCBI mammalian protein database, assuming that peptides were monoisotopic. Fixed modification was carbamidomethylation at cysteine residues, whereas variable modification was oxidation at methionine residues. Only one missed trypsin cleavage was allowed. Peptide mass tolerance was 1.2 Da, whereas fragmented mass tolerance was 0.6 Da. MS/MS ions scores >47 were considered statistically significant (significant hits). More details of Q-TOF MS/MS analysis are described in our previous study.15
Statistical analysis
To determine differences in absorbance at λ 570 nm in the competitive ninhydrin assay, unpaired Student's t-test was performed using SPSS software package for Windows (version 13.0) (SPSS; Chicago, IL). p values < 0.05 were considered statistically significant.Acknowledgements
We are grateful to Dr Mookda Pattrawarapan for her valuable advice and to Dr Rattiyaporn Kanlaya for her assistance in manuscript preparation. This work was supported by the Research Grant from Mahidol University, The Thailand Research Fund and Commission on Higher Education (to VT) and by the Thesis Scholarship from Faculty of Graduate Studies, Mahidol University (to PR). VT is also supported by “Chalermphrakiat” Grant, Faculty of Medicine Siriraj Hospital.References
- R. C. Walton, J. P. Kavanagh, B. R. Heywood and P. N. Rao, Biochim. Biophys. Acta, 2005, 1723, 175–183 CrossRef CAS.
- D. Sigmon, S. Kumar, B. Carpenter, T. Miller, M. Menon and C. Scheid, Am. J Kidney Dis., 1991, 17, 376–380 Search PubMed.
- Y. Ogawa, T. Miyazato and T. Hatano, J Am. Soc. Nephrol., 1999, 10(Suppl. 14), S341–S344 CAS.
- M. S. Schepers, E. S. van Ballegooijen, C. H. Bangma and C. F. Verkoelen, Kidney Int., 2005, 68, 1660–1669 CrossRef CAS.
- C. F. Verkoelen, J. C. Romijn, L. C. Cao, E. R. Boeve, W. C. De Bruijn and F. H. Schroder, J. Urol., 1996, 155, 749–752 CrossRef CAS.
- R. Selvam and P. Kalaiselvi, Urol. Res., 2003, 31, 242–256 CrossRef CAS.
- P. Latha, P. Kalaiselvi, P. Varalakshmi and G. Rameshkumar, Biochem. Biophys. Res. Commun., 2006, 345, 345–354 CrossRef CAS.
- M. B. Coulter-Mackie, Kidney Int., 2006, 70, 1891–1893 CrossRef CAS.
- D. P. Zhang, Z. Y. Wu, X. Y. Li and Z. X. Zhao, Plant Physiol., 2002, 128, 714–725 CrossRef CAS.
- S. K. Choi, M. Adachi and S. Utsumi, Biosci., Biotechnol., Biochem., 2002, 66, 2395–2401 CrossRef CAS.
- P. O'carra and S. Barry, Methods Enzymol., 1974, 34, 598–605 Search PubMed.
- J. B. Park, H. Z. Chae and Y. S. Kim, Korean Biochem. J., 1986, 19, 235–241 Search PubMed.
- M. N. Hwang and G. M. Ederer, J Clin. Microbiol., 1975, 1, 114–115 CAS.
- P. Sivakamasundari, P. Kalaiselvi, R. Sakthivel, R. Selvam and P. Varalakshmi, Clin. Chim. Acta, 2004, 347, 111–119 CrossRef CAS.
- R. Kanlaya, S. N. Pattanakitsakul, S. Sinchaikul, S. T. Chen and V. Thongboonkerd, Mol. BioSyst., 2010, 6, 795–806 RSC.
| This journal is © The Royal Society of Chemistry 2010 |