Combining EPR and ESI-MS/MS to study the reactivity of alkylthiols and cysteine towards 2-methyl-2-nitrosopropane (MNP)
Mathilde
Triquigneaux
,
Béatrice
Tuccio
and
Laurence
Charles*
Universités Aix-Marseille I, II & III—CNRS, UMR 6264, Laboratoire Chimie Provence, Spectrométries Appliquées à la Chimie Structurale, Campus Saint-Jérôme, Case 511, 13397, Marseille Cedex 20, France. E-mail: laurence.charles@univ-provence.fr; Fax: +33 491282897; Tel: +33 491288678
First published on 30th April 2010
Abstract
An approach combining EPR spectroscopy and electrospray ionisation mass spectrometry was employed to study the reactivity of two alkylthiols and of cysteine towards 2-methyl-2-nitrosopropane (MNP) in the presence of H2O2. Depending on the experimental conditions, various nitroxides could be EPR-detected, while their diamagnetic derivatives were structurally characterised after tandem mass spectrometry experiments. Sample dilution in methanol before electrospray ionisation was also found to generate methyl-hydroxylamine derivatives and could thus constitute a simple derivatisation process to stabilise hardly MS-detectable radical species. Upon white light irradiation, the three thiols tested led to alkoxynitroxides according to an inverted spin trapping process. On the other hand, only the reduced form of cysteine was found to react with MNP in the dark, yielding an alkylthionitroxide adduct via a Forrester–Hepburn mechanism. Besides a better understanding of the reactivity of thiols towards nitroso compounds, the results obtained underlined the particular behaviour of cysteine, suggesting that the use of a molecule as simple as MNP could allow a nitroxide moiety to be linked to a cysteinyl residue.
Introduction
Aminoxyl radicals, also called nitroxides, are stable or persistent paramagnetic species presenting a wide range of applications throughout material science, chemistry and biochemistry. Among them, site-directed spin labelling (SDSL), a technique in which a nitroxide is covalently linked to the thiol group of a cysteinyl residue, has emerged as a powerful method for investigating protein structure, dynamics and conformational changes.1–3 Once incorporated, the nitroxide spin label can be used as a probe, its motions being dictated by the protein local dynamics. Owing to their paramagnetic nature, electron paramagnetic resonance (EPR) spectroscopy is generally the technique of choice to study and characterise nitroxides, the spectra recorded being very sensitive to their mobility and to their environment. However, only poor structural information can be gained by EPR, nitroxides with similar structures generally yielding identical spectra. Thus, complementary approaches have to be used to fully characterise nitroxides. In previous studies, mass spectrometry (MS) has been employed to successfully overcome these limitations, using the MS mode to detect low quantities of both nitroxides and their diamagnetic derivatives, even in complex matrices, and performing tandem mass spectrometry (MS/MS) experiments for structural characterisation.4–6 The addition of reduced glutathione, a tripeptide containing one cysteinyl residue, on 2-methyl-2-nitrosopropane (MNP) in oxidative media was previously studied by the combination of EPR and mass spectrometry.7 It was then shown that different nitroxide adducts could be obtained depending on the experimental conditions. Upon white light irradiation, an alkoxynitroxide (i.e., an O-adduct) was EPR-detected, while the formation of an alkylthionitroxide (i.e., an S-adduct) was formed in the dark. Although these nitroxides could not be directly observed as cationised molecules in ESI-MS, their protonated hydroxylamine derivatives were detected in both cases and further characterised in MS/MS. Whatever the experimental conditions, the nucleophilic addition of reduced glutathione on MNP in an oxidative medium allowed to incorporate a nitroxide moiety in a tripeptide, and this interesting reactivity could open a new route to spin-label proteins. Therefore, a better understanding of the reactivity of various thiols with MNP could be useful.In the present study, the species generated upon reaction of MNP with two alkylthiols and with cysteine were investigated in order to scrutinise the influence of the thiol structure on the so-formed nitroxides. For this purpose, an EPR-MS(/MS) approach was employed, using the specificity and the sensitivity of EPR measurements to detect the nitroxides and performing MS and MS/MS experiments to fully elucidate their structure.
Experimental
Chemicals
Octanethiol (97%) and ammonium acetate (98%) were purchased from Sigma-Aldrich (St Louis, MO, USA). Propanethiol (98%), cysteine (99%) and 2-methyl-2-nitrosopropane (MNP) dimer (98%) were purchased from Acros, Thermo Fisher Scientific (Waltham, MA, USA) and H2O2 (30%) was from VWR (Fontenay sous Bois, France). Methanol (SDS, Peypin, France) and benzene (Sigma-Aldrich) were of the highest grade of purity commercially available and used as received.Deuterated methanol (CD3OD) was from Eurisotop (Saint-Aubin, France). Poly(ethylene glycol) with Mn 400 g mol−1 (Sigma-Aldrich) was used as an internal standard for accurate mass measurements.
Electron paramagnetic resonance
EPR assays were carried out at room-temperature in glass capillary tubes by using a computer-controlled EMX spectrometer (Bruker BioSpin, Silberstreifen, Germany) operating at X-band with 100 kHz modulation frequency and equipped with an NMR gaussmeter for magnetic field calibration. No EPR spectrum was ever detected when the capillaries were filled with pure solvent, proving that the glass signal did not alter the various spectra recorded. The instrument settings were as follows: non-saturating microwave power, 20 mW; modulation amplitude, 2 G; receiver gain, 1 × 106; time constant, 1.28 ms; scan time, 120 s; scan width, 100 G; 1 scan. Computer simulation of the EPR spectra was achieved using the program elaborated by Duling.8Mass spectrometry
High-resolution MS and MS/MS experiments were performed with a QStar Elite mass spectrometer (Applied Biosystems SCIEX, Concord, ON, Canada) equipped with an electrospray ionisation (ESI) source. In the positive ion mode, the capillary voltage was set at +5500 V and the cone voltage at +80 V. In this hybrid instrument, ions were measured using an orthogonal acceleration time-of-flight (oa-TOF) mass analyser. A quadrupole was used for selection of precursor ions to be further submitted to collision induced dissociation (CID) in a collision cell. In MS, accurate mass measurements were performed using two reference ions from a poly(ethylene glycol) internal standard, according to a procedure described elsewhere.9 The precursor ion was used as the reference for accurate measurements of product ion m/z ratio in MS/MS spectra. MS3 experiments were performed with a 3200 Q-TRAP mass spectrometer (Applied Biosystems SCIEX) equipped with an electrospray ionisation source operated in positive mode. The capillary voltage was set at +5500 V and the cone voltage at +80 V. Primary precursor ions generated in the ion source were selected in the quadrupole analyser and submitted to CID in a collision cell. Secondary precursor ions produced during collisions were selected and then fragmented in a linear ion trap. In both instruments, air was used as the nebulising gas (10 psi) whereas nitrogen was used as the curtain gas (20 psi) as well as the collision gas. Collision energy was set according to the experiments. Instrument control, data acquisition and data processing of all experiments were achieved using Analyst software (QS 2.0 and 1.4.1 for the QqTOF and the QqTrap instruments, respectively) provided by Applied Biosystems. Sample solutions were introduced in the ionisation source at a 5 µL min−1 flow rate using a syringe pump.Reactions between MNP and the thiols
The nitroxide adducts 1a, 2a and 3a (Scheme 1) were produced in benzene by mixing H2O2 (0.3%, v/v) and MNP (40 mmol L−1) with propanethiol, octanethiol or cysteine (5 mmol L−1), respectively. The system was then irradiated for 30 s using a tungsten filament 100 W lamp as the visible light source. The nitroxide adduct 4a was produced in water containing acetic acid (1%, final pH 4) by mixing MNP (40 mmol L−1) and cysteine (5 mmol L−1) for 20 min in the dark before adding H2O2 (0.3%, v/v). For each experiment, 300 µL of the reaction medium were prepared and an aliquot (ca. 30 µL) of the organic phase was analysed by EPR spectroscopy. MS samples were prepared by diluting the reaction medium (1/10, v/v) in a methanolic solution of ammonium acetate (3 mmol L−1).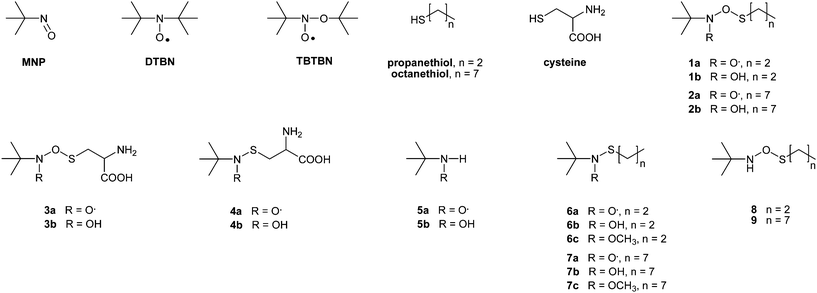 | ||
| Scheme 1 Chemical structure proposed for the thiol conjugates produced after reaction between propanethiol, octanethiol or cysteine with MNP. | ||
Results and discussion
MNP presents an n → π* transition in visible light, with a maximum absorption at 686 nm in water,10 and a MNP solution in benzene is pale blue. The photochemistry of MNP is well documented, and it has been shown that nitroxides could be formed after red light photolysis of this compound.11 In certain conditions, nitroxides can also be generated via the radical cations of the spin trap, formed by reaction between the excited state of MNP and an oxidant.12 Taking into account this particular behaviour of MNP, two different protocols were implemented in our study.Light-dependent reactions
In the first protocol, the experimental conditions consist of mixing MNP with a thiol in an oxidative medium, before irradiating the sample with white light. Regardless of the thiol tested (propanethiol, octanethiol or cysteine), the reaction with MNP gave rise to an EPR signal, identical to that obtained with reduced glutathione (Fig. 1a).7 It consisted of a large triplet due to a unique hyperfine coupling of the unpaired electron with nitrogen nucleus (aN = 27.1 G, g = 2.0056), the value of the hyperfine coupling constant (hfcc) aN being characteristic of an alkoxynitroxide.13–15 Structural assumptions could thus be proposed for these O-adducts, as illustrated in Scheme 1a by 1a, 2a and 3a for propanethiol, octanethiol and cysteine, respectively. However, a by-product formed after MNP irradiation in oxidative media, namely N-tert-butoxy-N-tert-butylnitroxide (TBTBN, Scheme 1), would generate almost identical EPR signal in benzene (aN = 26.6 G, g = 2.0058).7 In support to the presence of TBTBN in the medium, another well-known photo-degradation by-product of MNP, i.e. di-tert-butyl nitroxide (DTBN, Scheme 1), was also observed as a three line EPR (aN = 15.4 G, g = 2.0059). Consequently, mass spectrometry was used as an alternative technique to evidence the actual formation of the O-adducts 1a, 2a and 3a after the reaction between MNP and the thiol targets.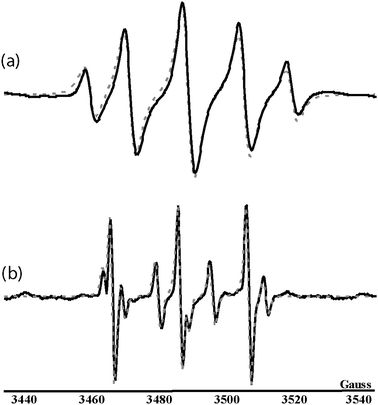 | ||
| Fig. 1 Experimental EPR signals (full lines) and superimposed simulation (dotted lines) of (a) O-adduct 3a (aN = 27.1 G, 32%) and DTBN (aN = 15.4 G, 68%) obtained from the reaction between cysteine and MNP in benzene, and (b) S-adduct 4a (aN = 18.3 G, 69%), H-adduct 5a (aN = 14.5 G, aH = 14.1 G, 21%) and DTBN (aN = 17.0 G, 10%) in aqueous medium (pH 4–5). | ||
The reaction media were diluted with methanol (1/10, v/v) before being submitted to electrospray ionisation, and the mass spectra were also scrutinised for the presence of protonated hydroxylamine derivatives in case the radical adducts could not be detected as charged species in mass spectrometry. The positive mode ESI-MS spectrum obtained with the propanethiol reagent did not show any signal associated to the ionisation of 1a whereas a peak at m/z 180.1 (m/z 180.1071: C7H18NO2S+, error: +1.7 ppm) could be assigned to the protonated hydroxylamine [1b + H]+ (Scheme 1). MNP, DTBN and TBTBN were not amenable to electrospray ionisation, probably because of their high volatility, and were not observed in the mass spectrum. CID of [1b + H]+ allowed the formation of five main product ions (Fig. 2) which formation could be accounted for, as described hereafter and illustrated in Scheme 2. Assuming a C7H18NO2S+ composition for the precursor ion to calibrate this MS/MS spectrum, accurate mass measurement indicated that m/z 124.1 (m/z 124.0453: C3H10NO2S+, error: +21.0 ppm) would arise from the elimination of a C4H8 neutral from the precursor ion. A second dissociation reaction of m/z 180.1 would produce two complementary product ions, depending on the location of the charge at the time of dissociation. It would consist of a 1,4-proton transfer from the OH group to the second oxygen atom, to give rise to either m/z 93.0 (m/z 93.0384: C3H9OS+, error: +16.1 ppm) after the elimination of MNP or protonated MNP at m/z 88.1 (m/z 88.0760: C4H10NO+, error: +3.4 ppm) if propane sulfenic acid is released as a neutral. The signal at m/z 57.1 (m/z 57.0706: C4H8+, error: +12.3 ppm) was assigned to the tert-butyl cation which would readily form upon protonation of the nitrogen atom and subsequent release of HO–NH–OC3H7 from the precursor ion. Finally, the low abundance product ion detected at m/z 58.1 (m/z 58.0658: C3H8N+, error: +12.1 ppm) would arise from the elimination of a formaldehyde molecule from the protonated MNP at m/z 88.1. Note that this result is the very first insight in the dissociation behaviour of [MNP + H]+ since this species could not be generated in the gas phase upon ESI of MNP due to its high volatility. All these product ions, and notably [MNP + H]+, along with the EPR data, would confirm that the signal observed at m/z 180.1 actually corresponds to [1b + H]+, that is, the protonated hydroxylamine derivative of a nitroxide resulting from the addition of the propanethiol-derived sulfenic acid on MNP.
![Proposed dissociation pathways of [1b + H]+ at m/z 180.1.](/image/article/2010/AY/c0ay00185f/c0ay00185f-s2.gif) | ||
| Scheme 2 Proposed dissociation pathways of [1b + H]+ at m/z 180.1. | ||
![ESI-MS/MS of [1b + H]+ at m/z 180.1 using a 15 eV collision energy (laboratory frame).](/image/article/2010/AY/c0ay00185f/c0ay00185f-f2.gif) | ||
| Fig. 2 ESI-MS/MS of [1b + H]+ at m/z 180.1 using a 15 eV collision energy (laboratory frame). | ||
A similar dissociation behaviour was observed for the protonated hydroxylamine 2b, detected at m/z 250.2 (m/z 250.1842: C12H28NO2S+, error: +2.8 ppm) in the mass spectrum of the reaction medium including octanethiol. Accurate mass data of the product ions detected in CID of m/z 250.2 are presented in Table 1 and validate the structure proposed for 2b.
| m/ztheo | Elemental composition | m/zexp | Error | |
|---|---|---|---|---|
| mDa | ppm | |||
| a I.S. = internal standard. | ||||
| [2b + H]+ | ||||
| 250.1835 | C12H28NO2S+ | I.S.a | — | — |
| 194.1209 | C8H20NO2S+ | 194.1226 | +1.7 | +8.8 |
| 163.1151 | C8H19OS+ | 163.1137 | −1.4 | +8.6 |
| 88.0757 | C4H10NO+ | 88.0774 | +1.7 | +19.3 |
| 58.0651 | C3H8N+ | 58.0648 | −0.3 | −5.2 |
| 57.0699 | C4H9+ | 57.0689 | −1.0 | −17.5 |
| [3b + H]+ | ||||
| 225.0904 | C7H17N2O4S+ | I.S.a | — | — |
| 169.0278 | C3H9N2O4S+ | 169.0292 | +1.4 | +8.3 |
| 152.0012 | C3H6NO4S+ | 152.0007 | −0.5 | −3.3 |
| 90.0913 | C4H12NO+ | 90.0921 | +0.8 | +8.9 |
| 57.0699 | C4H9+ | 57.0686 | −1.3 | −22.8 |
In contrast, the MS/MS pattern of [3b + H]+, the protonated hydroxylamine derived from cysteine, was slightly different. Indeed, CID of m/z 225.1 (m/z 225.0913: C7H17N2O4S+, error: +4.0 ppm) did not allow the formation of MNP, neither in its neutral nor in its protonated forms. However, m/z 225.1 was shown to dissociate via the elimination of tert-butene to produce m/z 169.0, or via the formation of the tert-butyl cation (m/z 57.1), as reported in the case of propanethiol and octanethiol O-adducts. The m/z 169.0 product ion could further eliminate NH3 to form a minor product ion at m/z 152.0. In addition, a dissociation reaction from the m/z 225.1 precursor ion would also produce m/z 90.1. This pathway would consist of a 1,7-proton transfer, inducing the loss of three neutrals, CO2, ethylenamine and SO. Accurate mass measurements of the main product ions of [3b + H]+ are summarised in Table 1. It should be noted that the dissociation pathways of [3b + H]+ are specific to the O-adduct of cysteine and does not present any similarity with the fragmentation of the protonated amino acid. More particularly, CID of [cysteine + H]+ was shown to give rise to the loss of CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S via a hydrogen transfer from the thiol group,16 which can no longer occur when this function is substituted. The lack of such an elimination during the dissociation of the protonated 3b would support the formation of a cysteine conjugate.
S via a hydrogen transfer from the thiol group,16 which can no longer occur when this function is substituted. The lack of such an elimination during the dissociation of the protonated 3b would support the formation of a cysteine conjugate.
To conclude, detailed information obtained from both EPR and MS/MS analyses allowed the validation of structural assumptions proposed for O-adducts formed with propanethiol, octanethiol and cysteine, respectively. More particularly, 1a, 2a and 3a nitroxides would be obtained from the inverted spin trapping reaction (Scheme 3), consisting in a prior oxidation of the nitroso compound into a radical cation before the addition of a sulfenic acid derived from thiol compounds.
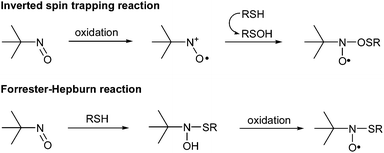 | ||
| Scheme 3 Formation of O- and S-adducts according to an inverted spin trapping reaction and a Forrester–Hepburn mechanism, respectively. | ||
Reactions occurring in the dark
The second experimental set-up consisted of mixing in the dark MNP with propanethiol, octanethiol or cysteine for 20 min before adding H2O2. Contrarily to what was observed with glutathione,7 no S-adduct was ever EPR-detected when experiments were conducted with either alkylthiols or cysteine. In the case of cysteine, however, a slight pH decrease (from 6–7 to 3–4) allowed the formation of the expected S-adduct 4a, as indicated by the three line EPR pattern shown in Fig. 1b (aN = 18.3 G, g = 2.0063). According to data previously published for various MNP/thiyl radical spin adducts,17,18 this aN value indicates the presence of a sulfur atom directly bonded to the nitroxide nitrogen. A four line minor EPR signal, with 1-2-2-1 relative intensities, was also observed. Its analysis revealed hyperfine couplings with a nitrogen and a β-hydrogen nucleus (aN = 14.5 G, aH = 14.1 G, g = 2.0056). According to literature data,19,20 this signal could be unambiguously assigned to the nitroxide 5a (see Scheme 1), probably formed after MNP reduction by the cysteine thiol function.MS and MS/MS experiments were performed to gain structural information on 4a and 5a. While the positive mode ESI-MS spectrum of the cysteine/MNP/H2O2 solution showed no signal corresponding to any form of 5a, probably due to volatility issues, a low abundance ion at m/z 209.1 could be assigned to the protonated hydroxylamine [4b + H]+. It is worth mentioning that a protonated molecule derived from a methylated form of the hydroxylamine was also observed by ESI-MS in our previous study concerning the reaction between reduced glutathione and MNP. In the present work, no peak was detected at the expected value m/z 223.1 for such a methylated species when cysteine was used in place of glutathione. CID of [4b + H]+ gave rise to the formation of two main product ions at m/z 191.1 and m/z 74.1, as shown in Fig. 3. Accurate mass measurements (m/z 191.0859: C7H15N2O2S+, error: +5.2 ppm) indicated that the m/z 191.1 product ion was formed after a water molecule elimination from the precursor ion. The elemental composition associated to m/z 74.1 (m/z 74.0977: C4H12N+, error: +17.5 ppm) suggests that this ion arises from the dissociation of m/z 191.1, which would have eliminated a 4-amino-1,2-oxathiolan-5-one neutral. Several additional minor product ions were also observed, most of which could be accounted for as primary fragments of the m/z 209.1 precursor ion. A combined loss of a 2-methyl-1-propene molecule and ammonia would generate the m/z 136.0 product ion (m/z 136.0084: C3H6NO3S+, error: +15.4 ppm) while m/z 90.1 (m/z 90.0915: C4H12NO+, error: +2.2 ppm) would correspond to the protonated hydroxylamine of MNP. Finally, consecutive dissociation of m/z 191.1 would allow the formation of the m/z 118.1 product ion (m/z 118.0695: C5H12NS+, error: +8.5 ppm) via the elimination of NHCH–CO2H. The fragmentation pattern proposed for [4b + H]+ is summarised in Scheme 4 and clearly indicates that 4a arose from the addition of the cysteine thiol function onto MNP following a Forrester–Hepburn reaction, generating a hydroxylamine further oxidised into nitroxide (Scheme 3).
![Proposed dissociation pathways of [4b + H]+ at m/z 209.1.](/image/article/2010/AY/c0ay00185f/c0ay00185f-s4.gif) | ||
| Scheme 4 Proposed dissociation pathways of [4b + H]+ at m/z 209.1. | ||
![ESI-MS/MS of [4b + H]+ at m/z 209.1 using a 10 eV collision energy (laboratory frame).](/image/article/2010/AY/c0ay00185f/c0ay00185f-f3.gif) | ||
| Fig. 3 ESI-MS/MS of [4b + H]+ at m/z 209.1 using a 10 eV collision energy (laboratory frame). | ||
In contrast to the case of reduced glutathione, formation of the S-adduct 4a only occurred in acidic conditions. Though the experiments were not performed at 25 °C but at room temperature, this could be due to the difference in redox potentials between glutathione (E°7.4 = −264 mV) and cysteine (E°7.4 = −247 mV),21,22 which would determine the relative concentration of the reactive reduced thiol and the unreactive oxidised sulfhydryl. Monitoring the redox equilibrium of cysteine in mass spectrometry showed that the m/z 241.1 protonated sulfhydryl species dominated at pH 6 (Fig. 4a) while it became negligible as compared to the m/z 122.1 protonated reduced form at pH 4 (Fig. 4b). These results strongly suggest that only the reduced thiol form of cysteine reacted with MNP to yield the S-adduct 4a.
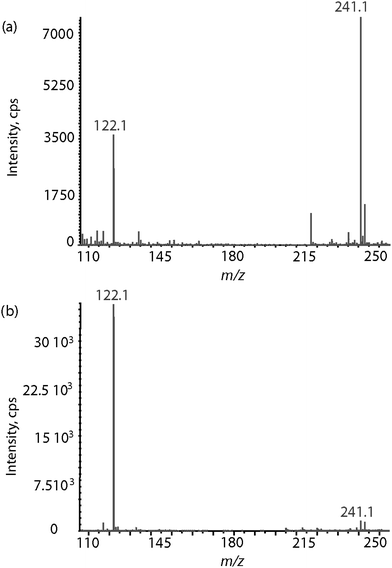 | ||
| Fig. 4 ESI mass spectra of a cysteine methanolic solution (a) at pH 6 and (b) at pH 4. The protonated reduced cysteine is detected at m/z 122.1 while the protonated oxidized cysteine is detected at m/z 241.1. | ||
Although nitroxide S-adducts were never EPR-observed from alkylthiols, the formation of related diamagnetic species was investigated by mass spectrometry. MS analysis of the propanethiol/MNP/H2O2 system in the positive ion mode revealed the protonated hydroxylamine [6b + H]+ (m/z 164.1096: C7H18NOS+, error: −4.9 ppm), and the protonated O-methyl hydroxylamine [6c + H]+ (m/z 178.1263: C8H20NOS+, error: +1.7 ppm). The reaction between octanethiol and MNP gave rise to similar results, with the detection of [7b + H]+ (m/z 234.1881: C12H28NOS+, error: −2.1 ppm) and [7c + H]+ (m/z 248.2031: C13H30NOS+, error: −4.8 ppm). Tandem mass spectrometry was then performed to validate these structural assignments. CID of [7b + H]+ at m/z 234.2 led to the formation of a main product ion at m/z 178.1. Accurate mass measurements (m/z 178.1247: C8H20NOS+, error: −7.3 ppm) indicated this ion was formed after the elimination of a C4H8 neutral from the precursor ion. Moreover, the complementary tert-butyl cation was also observed at m/z 57.1 (m/z 57.0693: C4H9+, error: −10.5 ppm). However, accurate mass measurements suggested that the m/z 161.1 product ion would result from the release of C4H11N from the precursor ion, which is hard to envisage from [7b + H]+. In contrast, such a dissociation pathway would readily occur from the protonated isomeric form 9 (Scheme 1). Although such a structure is unusual, other stable N-(alkylthio-oxy)-alkylamines have been described in the literature.23–25 A similar conclusion was reached from MS/MS data analysis of m/z 164.1, assigned to the protonated hydroxylamine derivative of the nitroxide obtained when using propanethiol. The same C4H11N neutral release is suggested by accurate mass measurement but would be more realistic to occur from [8 + H]+ rather than [6b + H]+.
However, peaks detected at m/z 178.1 and m/z 248.2 in the MS spectra obtained from reaction media containing respectively propanethiol and octanethiol could be assigned to O-methyl hydroxylamine protonated forms of 6c and 7c. Indeed, CID of m/z 248.2 gave rise to a unique product ion at m/z 192.1 (m/z 192.1419: C9H22NOS+, error: +1.0 ppm) formed after elimination of a C4H8 molecule, and further dissociation of this product ion mainly yielded m/z 80.0 after the release of octane, as evidenced in MS3 experiments. Similarly, the MS/MS filiation m/z 178.1 → m/z 122.16 → m/z 80.0 revealed the successive elimination of isobutene and propene. Considering that 6c and 7c arose from the methylation of respectively 6b and 7b, either in the liquid or in the gas phase, additional experiments involving H/D exchanges were conducted to elucidate this process. In a first experiment, further referred as the CD3OD/CH3OH sequence, MNP and propanethiol were reacted in deuterated methanol prior to dilution with methanol. Positive ion mode electrospray of this solution generated a main m/z 181.1444 ion, which accurate mass value corresponds to C8H17D3NOS+ (error: −2.4 ppm). These results indicate that the m/z 181.1 ion obtained from the CD3OD/CH3OH sequence was the protonated homologue of 6c with R = OCD3 (Scheme 1). A second experiment involved a CH3OH/CD3OD sequence, i.e. a reaction between MNP and propanethiol in methanol before using deuterated methanol as diluting solvent. In this case, an ion was detected at m/z 179.1326 with the associated elemental composition C8H19DNOS+ (error: +1.7 ppm). Results obtained in the CH3OH/CD3OD experiment would thus imply a methylated nitroxide R = OCH3 moiety and the cationisation of the so-formed molecule with a D+ during electrospray. These results unambiguously demonstrate that methylation exclusively occurred in the liquid phase during the methanol dilution step, implemented for the purpose of ESI-MS analysis. Consequently, this reaction should be considered as an efficient and simple derivatisation process to stabilise hardly detectable radical species, thereby allowing full MS/MS structural characterisation.
EPR analyses never revealed the presence of the S-adducts 6a and 7a, but their formation after nucleophilic addition of propane- or octane-thiol on MNP was suggested by the detection of [6c + H]+ and [7c + H]+. In such a Forrester–Hepburn mechanism, the hydroxylamines 6b and 7b could be considered either as nitroxide precursors, formed before the oxidation step, or as nitroxide derivatives, obtained after a reductive process. When additional experiments were performed in the absence of H2O2, 6c and 7c were still detected as protonated molecules in mass spectrometry, clearly revealing the formation of hydroxylamines 6b and 7b as nitroxide precursors. On the other hand, increasing the oxidant H2O2 concentration up to 20% never allowed the EPR detection of 6a and 7a. Although a nitroxide lifetime strongly depends on its environment, these results suggest a fast conversion of 6a and 7a into the corresponding hydroxylamines in the experimental conditions used, while longer-lived paramagnetic species were obtained from cysteine thiols.
Conclusion
The EPR/MS approach, previously used in conjunction with spin trapping to identify short-lived free radicals, was shown here to be an efficient tool to characterise nitroxides obtained after nucleophilic addition of various thiols on MNP, as well as their diamagnetic derivatives. The obtained results allowed a better understanding of the reactivity of thiols towards MNP. They notably underline the particular behaviour of the cysteine thiol function. The stability of thio-adducts, in particular S-adducts, was also shown to be logically influenced by the structural environment of the nitroxide function, the species obtained after reaction of cysteine on MNP being rather persistent in our experimental conditions. Besides the interest of the EPR/MS method for mechanism elucidation in radical chemistry, all these results suggest that the use of a molecule as simple as MNP could allow a nitroxide moiety to be linked to a cysteinyl residue, yielding paramagnetic species persistent enough for structural analysis. Of course, validation of this method for protein spin labelling requires further studies that fall out of the scope of this paper. In particular, an extensive kinetic study would permit to evaluate the stability of the nitroxides obtained after reaction between MNP and either cysteine or reduced glutathione. The process should also be improved to obtain spin-labelled compounds in mild conditions in order to avoid protein denaturation.Acknowledgements
L. Charles acknowledges support from Spectropole, the Analytical Facility of Aix-Marseille University, by allowing a special access to the instruments purchased with European Funding (FEDER OBJ2142-3341).References
- C. S. Klug and J. B. Feix, Methods and Applications of Site-Directed Spin Labeling EPR Spectroscopy, in Biophysical Tools for Biologists, Elsevier, Academic Press Inc, San Diego, 2008, vol. 1, pp. 617–658 Search PubMed.
- A. Czogalla, A. Pieciul, A. Jezierski and A. F. Sikorski, Acta Biochim. Pol., 2007, 54, 235–244 CAS.
- G. E. Fanucci and D. S. Cafiso, Curr. Opin. Struct. Biol., 2006, 16, 644–653 CrossRef CAS.
- I. El Hassan, L. Charles, R. Lauricella and B. Tuccio, New J. Chem., 2008, 32, 680–688 RSC.
- B. Tuccio, R. Lauricella and L. Charles, Int. J. Mass Spectrom., 2006, 252, 47–53 Search PubMed.
- M. Triquigneaux, L. Charles, C. André-Barrès and B. Tuccio, Org. Biomol. Chem., 2010, 8, 1361–1367 RSC.
- M. Triquigneaux, B. Tuccio, R. Lauricella and L. Charles, J. Am. Soc. Mass Spectrom., 2009, 20, 2013–2020 CrossRef CAS.
- D. R. Duling, J. Magn. Reson., Ser. B, 1994, 104, 105–110 CrossRef CAS.
- L. Charles, Rapid Commun. Mass Spectrom., 2008, 22, 151–155 CrossRef CAS.
- A. Calder, A. Forrester and S. Hepburn, Org. Synth., 1972, 52, 77–82 CAS.
- L. Eberson, J. Lind and G. Merenyi, J. Chem. Soc., Perkin Trans. 2, 1994, 1181–1188 RSC.
- (a) F. Broekhoven, T. Bolsman and T. De Boer, Recl. Trav. Chim. Pays-Bas, 1977, 96, 12–16 CAS; (b) K. Makino, N. Suzuki, F. Moriya, S. Rokushika and H. Hatano, Radiat. Res., 1981, 86, 294–310 CAS.
- M. Lucarini, G. F. Pedulli, A. Alberti and M. Benaglia, J. Am. Chem. Soc., 2002, 114, 9603–9607.
- S. W. Mao and L. Kevan, Chem. Phys. Lett., 1974, 24, 505–507 CrossRef CAS.
- F. P. Sargent and E. M. Gardy, J. Phys. Chem., 2002, 80, 854–856 Search PubMed.
- R. A. J. O'Hair, M. L. Styles and G. E. Reid, J. Am. Soc. Mass Spectrom., 1998, 9, 1275–1284 CrossRef CAS.
- C. C. Felix, K. Reszka and R. C. Sealy, Photochem. Photobiol., 1983, 37, 141–147 CrossRef CAS.
- J. Jing, W. Longmin and Z. Ziyi, Magn. Reson. Chem., 2002, 40, 346–352 CrossRef CAS.
- C. F. Chignell, B. Kalyanaraman, R. H. Sik and R. P. Mason, Photochem. Photobiol., 1981, 34, 147–156 CAS.
- F. P. Sargent and E. M. Gardy, Can. J. Chem., 1976, 54, 275–279 CAS.
- F. Q. Schafer and G. R. Buettner, Free Radicals Biol. Med., 2001, 30, 1191–1212 CrossRef CAS.
- J. H. Suh, R. Kim, B. Yavuz, D. Lee, A. Lal, B. N. Ames and M. K. Shigenaga, J. Chromatogr., B: Biomed. Appl., 2009, 877, 3418–3427 CrossRef CAS.
- C. Brown, R. F. Hudson and K. A. F. Record, J. Chem. Soc., Perkin Trans. 2, 1978, 822–828 RSC.
- B. Mile, C. C. Rowlands, P. D. Sillman and M. Fildes, J. Chem. Soc., Perkin Trans. 2, 1992, 1431–1437 RSC.
- R. Sustmann, W. Sicking and R. Huisgen, J. Am. Chem. Soc., 2002, 117, 9679–9685.
| This journal is © The Royal Society of Chemistry 2010 |