Comparative investigation of PdR by usual and ultrafine atomic force microscopy
Yuri D.
Ivanov
*a,
Pavel A.
Frantsuzov
a,
Victor A.
Bykov
b,
Stanislav P.
Besedin
c,
Gaston Hui
Bon Hoa
d and
Alexander I.
Archakov
a
aInstitute of Biomedical Chemistry, Pogodinskaya st. 10, Moscow, 119121, Russia. E-mail: yurii.ivanov@rambler.ru; Fax: +7 499-245-08-57; Tel: +7 499-246-37-61
bNT-MDT, Moscow, Russia
cKurchatov Institute, Moscow, Russia
dINSERM U779, 78 rue du General Leclerc, Le Kremlin Bicetre, France
First published on 23rd April 2010
Abstract
Atomic force microscopy is one of the most perspective methods for determination of the structure of proteins and their complexes. The vertical resolution of this method is about 0.1 nm, which is close to X-ray resolution. At the same time, the lateral resolution, determining by broadening effect of a standard AFM probe, is not very high—about 20–50 nm, depending on probe geometry. Naturally, the probe tip broadening effect leads to substantial enhancement of measured protein volume. In this study, a comparative analysis of sizes of the protein putidaredoxin reductase (PdR) obtained by the use of two AFM probe types, standard and supersharp, was undertaken. Usage of standard probes enabled to correctly measure the height of PdR while the volume of this protein was measured with considerably (more than one order) enhancement. It was shown that application of supersharp AFM probes results in the lowering of measured protein height; at the same time, the measured protein volume is more exact and appears to be close to RSA data. Therefore, to obtain exact data on protein volume and height, these two parameters should be measured by use of both supersharp probes and standard geometry probes.
Introduction
Determination of the structure of proteins and their complexes is one of the key problems of proteomics. Information on proteins in individual and complexed states is of fundamental importance in view of the major role of protein–protein interactions in cell function. The commonly adopted methods for structural analysis of proteins and protein complexes are based on such popular technologies as X-ray and nuclear magnetic resonance (NMR) analyses.1–3 In recent years, a fast-developing technique, AFM, is finding an increasing application in studies of biological objects4,5—owing to its ability to examine these objects in near-native conditions. Additional advantages of this technique are: the simplified procedure of sample preparation, the possibility to register and visualize proteins in both the individual and complexed states and the ease of data interpretation. The vertical resolution of AFM is very high—about 0.1 nm.6 However, when measuring lateral sizes, one comes up against two problems: probe broadening effect and sample shift under the influence of the scanning probe. If the AFM is equipped with the usual (in further text, standard) probe whose radius of curvature R is about 10 nm, the lateral sizes of isolated protein molecule, adsorbed on AFM support (at room temperature, atmospheric pressure and ordinary humidity), are about 5–10 times greater (depending on the probe's manufacturer) than the ones obtained from X-ray studies.7–10 This leads to considerable overestimation of the measured lateral sizes of protein molecules. In view of this, the volumes of these molecules, as measured by AFM, appear to be grossly enlarged compared to appropriate X-ray values. Researchers tried to solve this problem by two ways: (a) by restraining the motility of isolated proteins on support under the probe force; and (b) by lowering the probe's radius of curvature. The restraining of protein motility on support may be achieved by temperature decrease—as was demonstrated in observation over the Y-form of IgG on mica.11 However, this approach is complicated by difficulty in obtaining stable images—due to the influence of adsorbed and frozen inclusions on the probe and sample surfaces. The restraining of protein motility at room temperature (i.e. without freezing) is achieved by elimination of water layer adsorbed from air onto AFM support.12 For this purpose, the relative humidity was decreased to 30%.Another approach to increasing lateral resolution is based on the usage of probes with lowered radius of curvature. Such recently developed probes have the characteristic radius of curvature of about 1 nm. Application of such probes (based on carbon nanotubes) made it possible to achieve ultrafine resolution of the Y-form of IgG adsorbed onto freshly cleaved mica.13 However, high-quality images are not obtainable for all proteins because of enhanced motility of protein under the influence of “nanotube” probes. In this study we examined the possibility of AFM visualization of protein (whose motility was restrained by removal of water adsorbed on AFM support in vacuum) by use of supersharp probes. To obtain, by use of supersharp AFM probe, a high-quality image of protein, the protein sample must tightly adhere to the support surface—in order to withstand the probe force. The choice of vacuum regime of measurement is explained by the following considerations. In Ref. 12 it was noted that the layer of adsorbed water hampers obtaining high-quality images of proteins. The authors recommended removing the adsorbed water layer by drying the AFM support in a dessicator with dehydrated silicagel for 1 week. However, 1–2 h after AFM supports were removed from the dessicator and exposed to air, the quality of images was deteriorated due to readsorbtion of water from air. In our study the approach based on removal of adsorbed water layer by vacuum pumping was used. To prevent repeated readsorbtion of water onto the AFM support, the PdR visualization was carried out in vacuum using vacuum NTEGRA Aura microscope (NT-MDT, Russia). Data on protein heights and volumes, obtained by use of supersharp vs. standard probes, were compared. As the subject of study, the protein putidaredoxin reductase (PdR) was chosen. PdR is a protein of the electron-transport chain of the cytochrome P450cam-containing monooxygenase system involved in camphor metabolism.14,15 The choice of PdR for our studies was determined by the already solved crystal structure of this protein. It has a pyramidal form and measures 2.5 nm × 4.3 nm × 6.5 nm with a volume of about 20 nm3.16 The knowledge of PdR's structure makes it possible to compare AFM-made measurements with appropriate X-ray data.
Experimental
Chemicals
1-Ethyl-3-(3-dimethyl-aminopropyl)carbodiimide (EDC), N-hydroxysuccinimide (NHS), Tris were from Sigma. All other chemicals were from Reakhim (Moscow, Russia). Ultrapure water was obtained using the Milli-Q system (Millipore, MA, USA).Preparation of proteins
Putidaredoxin reductase was expressed in E.coli strain TB, isolated and purified to homogeneity as described elsewhere.17 Homogeneity of PdR was verified by SDS.Analytical measurements
Absorption spectra of the PdR was measured on an Agilent Model 8453 spectrophotometer at 25 °C. The concentration of purified PdR was determined from their absorption spectrum with the extinction coefficient 10 mM−1 cm−1 at λ = 454 nm.18AFM apparatus
AFM-measurements were conducted on the multimode NTEGRA Aura atomic force microscope (NT-MDT, Moscow, Russia).AFM measurements were conducted using two probe types: supersharp probes with radius of curvature of about 1–3 nm and probes of standard curvature with the radius of curvature 10–30 nm. As supersharp probes, NSG01_DLC microprobes (NT-MDT, Russia) with a typical resonance frequency 115–190 kHz were used.
As standard probes, NSG10 microprobes (NT-MDT, Russia) with a typical resonance frequency 190–325 kHz were used.
As support was used the negatively charged mica (grade V4) surface (SPI, USA).
Sample preparation
Two schemes of PdR immobilization on AFM support were used: non-covalent and covalent ones.For noncovalent immobilization of protein, 5 μl of 0.66 μM solution of PdR in 50 mM Tris buffer, containing 200 mM KCl, pH 7.4, were deposited onto the mica surface and left for 2 min. Then each sample was rinsed with ultrapure distilled water and dried in airflow.
Covalent immobilization of PdR across the carboxy groups onto the surface of aminosilanized mica was conducted using EDC according to Ref. 19. For this purpose, aminosilanized mica was incubated in 50 mM K-phosphate buffer (KP), pH 7.4, containing PdR (1 μM) and (EDC/NHS = 0.08![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.02 M) for 2 min whereupon non-covalently adsorbed proteins were washed off from the surface of the AFM-chip with the use of deionized water; then the support was dried in airflow.
:0.02 M) for 2 min whereupon non-covalently adsorbed proteins were washed off from the surface of the AFM-chip with the use of deionized water; then the support was dried in airflow.
AFM-measurements
To conduct AFM measurements in vacuum, the surface with immobilized protein was placed into the AFM-chamber. Prior to measurements the chamber was vacuumized to the residual pressure 10−2 Torr.AFM measurements in air were carried out at 60% relative humidity and T = 25 °C
All AFM experiments were carried out in a tapping mode. The scanning step was 1 nm. The scan size was 1 μm ×1 μm in case of supersharp probe and 3 × 3 μm in the case of the standard probe.
In control experiments, an appropriate buffer mixture without proteins was deposited onto mica and imaged. Randomly distributed contaminations in control measurements were less than 0.8 nm high.
Data analysis
AFM images' heights and volumes were calculated in automatic mode using GRF software (IBMC RAMS, Russia, www.soft.ibmc.msk.ru). Not less than 10 experiments were carried out with each type of probe (standard and supersharp), the number of objects was not less than 600 in each experiment.The AFM-object distribution with heights ρ(h) was calculated as follows:
| ρ(h) = (Nh/N) × 100% | (1) |
The volumes of AFM-imaged objects were calculated using GRF software as V = Σ(Sihi), where Si is a local area of the object, depending on the scanning step value and hi is the respective local height.
The AFM-objects distribution with heights and volumes ρ(h,V) was calculated as follows:
| ρ(h,V)=(Nh,V/N)*100% | (2) |
Results
AFM visualization of PdR molecules with standard probes
The control experiments on visualization of PdR, non-covalently adsorbed onto mica, were conducted in vacuum using AFM with standard probes. Fig. 1 presents standard AFM images of PdR molecules. Since in these experiments the images of molecules are markedly broadened, we have resorted to comparative analysis of distributions of imaged objects with heights.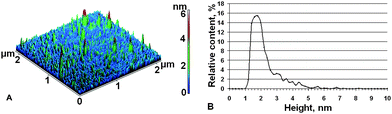 | ||
| Fig. 1 Vacuum AFM (standard probe) images of PdR adsorbed on mica. Experimental conditions were: 0.66 μM PdR in (50 mM Tris + 200 mM KCl) buffer (pH = 7.4, t = 25 °C) were deposited on mica surface for 2 min, rinsed with deionized water and dried in airflow. Tapping mode (A) and density of distribution ρ(h) (B), calculated from Fig. 1 (A). | ||
Approximation of experimental dependence (1) was carried out by the root mean square method using the ρ(h) function:
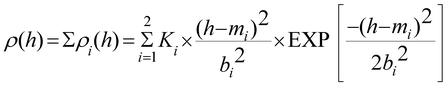 | (3) |
The analysis of approximation of the experimental curve based on χ2-criterion shows that this curve was poorly approximated by one-exponential function (because in this case χ2 = 1.7>1) and well approximated by the sum of two exponents (χ2 = 0.7<1). In view of this, the latter approximation variant was chosen (see Table 1). In the same table the contribution of each exponent to ρ(h) distribution are presented. As seen from Table 1, ρ(h) is characterized by the sum of two distributions: (a) the distribution of AFM images with the height hmax1 = 1.6 ± 0.2 nm (whose full width at half maximum, FWHM1 = 0.7 ± 0.2 nm), corresponding to the maximum number of imaged objects; and (b) the distribution of the AFM images with the height hmax2 = 2.5 ± 0.2 nm (whose FWHM2 = 2.0 ± 0.2 nm), corresponding to the maximum number of that objects. It is reasonable to suggest that the distribution of 1st type ρ1(h) with the smaller hmax1 corresponds to the distribution of PdR monomers while the distribution of 2nd type ρ2(h) with the larger hmax2 corresponds to the distribution of PdR oligomers. The integral share of oligomers, described by distribution ρ2(h) makes up α = 45 ± 9%.
| Objects | h max1/nm | V max1/nm3 | h max2/nm | V max2/nm3 | Share of objects (%) |
|---|---|---|---|---|---|
| Monomers | 1.6 ± 0.2 | 600 ± 200 | 55 ± 10 | ||
| Aggregates | 2.5 ± 0.2 | 2400 ± 600 | 45 ± 9 |
Lateral sizes of imaged PdR monomers were in the order of 30–80 nm, with the most probable value of about 40 nm. Assuming that the characteristic size of protein D0 is roughly equal to the intermediate value between the lateral sizes 6.5 nm and 4.3 nm, it may be concluded that the broadened sizes of PdR images obtained by use of standard AFM probe are about 7 times greater than the ones obtained from X-ray analysis.
The volumes of imaged monomers (Table 1) were characterized by Vmax1 = 600 ± 200 nm3 for objects with hmax1 = 1.6 ± 0.2 nm, while the volumes of imaged aggregates had Vmax2 = 2400 ± 600 nm3 for objects with hmax2 = 2.5 ± 0.2 nm. One can see that the volume of PdR monomer is ∼30 times greater than the appropriate volume V = 20 nm3, which is calculated from X-ray data.
Let us estimate the measurement error for molecular volume for revelation of contribution from measurement error for height and lateral sizes. The volume of imaged protein molecule may be calculated as:
| V = ΣhiSi(hi) | (4) |
| δSo = ΔSo/So = [(D + ΔD)2 – D2]/D2 = 2ΔD/D+ (ΔD/D)2 = 2δD+ δD2 | (5) |
| δV = ΔV/V = ΔSo/So + Δh/h = δSo + δh = (2δD+ δD2) + δh | (6) |
 | (7) |
Thus, usage of standard probes does not enable to correctly measure the PdR volume, which was measured with considerable overestimation.
AFM visualization of PdR molecules using supersharp probes
Presented in Fig. 2 (A) are the images of adsorbed-on-mica PdR molecules obtained by AFM with supersharp probes in vacuum. One can see that the imaged objects thus obtained are structurally different: some take an ellipsoidal form, others emerge as two-peak structures and still others as more complex structures. It was suggested that images of various forms correspond to various aggregated states of PdR. Enhanced lateral resolution enables calculation the volumes of imaged objects corresponding to PdRs of various aggregation states. Given below is the analysis of AFM images obtained in vacuum with the aid of supersharp probes. | ||
| Fig. 2 Vacuum AFM (supersharp probe) images of PdR adsorbed on mica. Experimental conditions were: 0.66 μM PdR in (50 mM Tris + 200 mM KCl) buffer (pH = 7.4, t = 25 °C) were deposited on mica surface for 2 min, rinsed with deionized water and dried in airflow. Tapping mode. Arrows 1, 2, 3 and 4 indicate, respectively, the images of PdR monomer, dimer, trimer and molecular aggregates of higher order (A); density of distribution ρ(h, V) (B), calculated from Fig. 2 (A); density of distribution ρ(h) (C), calculated from Fig. 2 (A). | ||
Distribution of images with heights and volumes ρ(h, V) calculated from eqn (2) is presented in Fig. 2 (B).
Objects, corresponding to this distribution, may be conventionally divided into 4 groups (see Table 2): (1) objects with heights residing in the interval h = 1.0÷2.0 nm with hmax = 1.2 ± 0.1 nm corresponding to the distribution maximum of objects with heights. The volume of objects, corresponding to this hmax, amounts to Vmax = 15 ± 4 nm3; (2) objects of double volume with heights in the interval h = 1.0÷2.0 nm, hmax = 1.4 ± 0.1 nm and Vmax = 35 ± 10 nm3; (3) complex objects of triple volume with heights in the interval h = 1.2÷2.4 nm, hmax = 1.6 ± 0.1 nm and Vmax = 60 ± 15 nm3 and, also, complex objects with heights in the interval h = 1.4÷2.2 nm, hmax = 1.6 ± 0.1 nm and Vρmax = 80 ± 15 nm3; (4) complex objects with heights in the interval h = 1.6÷3.0 nm, hmax = 1.8 ± 0.1 nm and Vmax = 115 ± 20 nm3.
| Objects | h max/nm | V max/nm3 | Aggregation degree | Share of objects (%) |
|---|---|---|---|---|
| Ellipsoidal | 1.2 ± 0.1 | 15 ± 4 | monomers | 31 ± 7 |
| Objects with double volume | 1.4 ± 0.1 | 35 ± 10 | dimers | 11 ± 3 |
| Objects with triple and 4-fold volume | 1.6 ± 0.1 | 60 ± 15 ÷ 80 ± 15 | trimers-tetramers | 28 ± 5 |
| Other objects | 1.8 ± 0.1 | 115 ± 20 | higher order aggregates | 30 ± 7 |
Comparison of volumes Vmax of AFM-imaged objects in group (1), presented in Table 2, with the volume of PdR monomers (20 nm3) from X-ray data shows that objects with minimal sizes, i.e. those residing in group (1) correspond to PdR monomers accounting for 31%±7% of the total number of objects. Lateral sizes of imaged PdR monomers were in the order of 7–11 nm, with the most probable value ∼8 nm. Assuming that the characteristic diameter of the protein D0 ∼ 5.4 nm, it may be concluded that the image of PdR monomer is broadened by the supersharp AFM probe by 2–3 nm compared to X-ray data.
Objects in group 2 with the volume Vmax being twice larger than the volume of monomers apparently correspond to imaged dimers accounting for 11 ± 3% of the total number of objects. Objects in group (3) with Vmax exceeding by three-fold the volume of monomers and apparently corresponding to trimers as well as objects with Vmax exceeding by about four-fold the volume of monomers and apparently corresponding to tetramers are united in one group in view of the fact that their volumes are within the experimental error. Integral share of these objects accounts for 28 ± 5%. Objects in group (4) correspond to aggregates of higher orders, whose share amounts to 30 ± 7%. Thus, oligomers integral share makes up ∼69%.
The height of group (1) imaged objects corresponding to monomers has the value of hmax = 1.2 ± 0.1 nm which is considerably (twice) less than the height of PdR from X-ray data (2.5 nm). It should be noted, that the height of PdR monomer, determined by AFM with standard probe (1.6 nm), is closer to appropriate X-ray data (2.5 nm). To minimize the influence of probe on sample, the minimal possible force was applied. For this purpose, prior to measurements on NTEGRA Aura AFM in vacuum, we have estimated the dependence of the probe's oscillation amplitude on the distance between probe and sample and have set the oscillation amplitude to the minimally possible level (in the order of 10 nm). With setting the lesser oscillation amplitude, i.e. with diminishing the probe force, the unstable measurement regime was established.
The lowered value of PdR height may be suggested to be due to the motility of the PdR molecule under the supersharp probe force, or to the spreading of PdR molecules, or else to their shrinkage by AFM probe, or some other, yet unknown causes.
To assess the influence of the motility effect, PdR was covalently immobilized onto mica. It was found that PdR images obtained are similar to those of the non-covalently adsorbed protein with the same ratio of monomeric to oligomeric forms. Therefore, height lowering is not associated with the protein's motility.
Using eqn (5) we may estimate the measurement error for the protein's volume obtained with the supersharp probe. Assuming that the characteristic diameter (lateral size) of imaged monomeric protein D = 8 nm and its height h = 1.2 nm, whereas the X-ray—measured protein size D0 = (6.5 + 4.3)/2∼5.4 nm and its height h0 = 2.5 nm, we derive from eqn (5) that the measurement error for the protein's volume is:
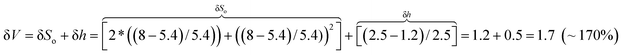 | (8) |
Of note, the measurement error for the protein's volume, which was calculated from comparison of data obtained with supersharp probe (15 nm3) and X-ray data (20 nm3), is ((20 nm3 – 15 nm3)/20 nm3) × 100% = 25%. This value is much less than the 170% value obtained from eqn (8). Such underestimation of actually measured volume may occur for two reasons: (a) the calculation derived from eqn (8) was based on the assumption that the lateral size of the protein molecule is roughly equal to its diameter while in reality upon measuring the volume we have taken into account the value of the function S(h), hence the underestimation of the volume; (b) measurement of the imaged object's volume was accompanied by the decrease of its height under the probe force which in reality led to the decrease (i.e. error) of measured volume—that is, the contribution of the second member to eqn (8) was not positive but negative. The latter reason appears to be essential since its non-account may lead to wrong interpretation of results obtained.
Thus, the measurement of protein volume leads to an error, which is connected with the interrelationship of two factors acting in opposite directions: (a) the factor increasing the measured volume at the cost of lateral broadening and (b) the factor that decreasing the volume through decreasing of the height, which leads to the negative contribution to the measurement error. Therefore the actual volume decreases and accounts closer to the one measured by X-ray analysis.
The difference between data on sizes of protein obtained by two methods (X-ray analysis and AFM with supersharp probes) can possibly be explained by differences in experimental conditions and in data interpretation. Thus, in X-ray experiments the protein gets crystallized, while upon AFM experiments the protein is visualized not in crystal state but as individual molecules. Besides in AFM the height of every molecule is determined directly after which the distribution ρ(h) is calculated whence the protein height is determined as the height of objects whose share is maximal. X-ray analysis provides information on the size of the protein molecule from diffraction picture of protein crystal.
Thus, AFM with supersharp probe enables to correctly measure the protein molecule's volume whose value appears to be close to X-ray data while the protein molecule's height is measured incorrectly—with underestimation of its value. Naturally, the need to compare the results obtained by use of AFM with supersharp and standard probes becomes apparent.
Now, compare the measurement errors for protein volume upon usage of standard and supersharp probes. As seen from eqn (6) and (8), the error arising upon protein volume determination by use of supersharp probe makes up ∼170% while the appropriate error with standard probe amounts to ∼5430%. That is, in passing from standard to supersharp probe, the measurement error for the PdR monomer's volume is lowered by an order.
Of note, when measured in air at ∼60% humidity, the non-covalently immobilized PdR molecules shifted over the surface under the supersharp AFM probe force—which did not make it possible to obtain PdR images of satisfactory quality. By contrast, AFM measurements carried out in air by use of standard probes made it possible to obtain high-quality images of PdR adsorbed onto mica surface. It appears therefore that in passing from standard to supersharp probes the motility of protein on support is increased under the probe force. As is known, upon measurements in air at relative humidity >45%, mica gets covered with water layer.17 Weak adhesion of protein molecules on AFM support is probably due to the adsorbed-on-support water layer.12 This means that during AFM measurements in air with supersharp probe the protein undergoes the higher pressure than upon usage of standard probe, which leads to PdR molecule's shift along the support.
Thus in passing from standard AFM probe to the supersharp one, it is possible to lower (by more than an order) the measurement error for protein volume. At the same time, the measurement of height by use of standard probe appears to be more correct than by use of supersharp probe. Therefore, to obtain the protein's height value, it is expedient to use standard probes while in obtaining the lateral size values the supersharp probes should apparently be preferred. Therefore, combined usage of AFM with supersharp and standard probes provides more correct information on protein heights and volumes than does the usage of only one probe type.
Acknowledgements
This work was supported by FASI contract N 02.552.11.7060. Support was also provided by RFBR Grant # 09-04-12113-OFR_M and Special RF Programme: Proteomics in medicine and biotechnology.References
- V. Yu. Kuznetsov, E. Blair, P. J. Farmer, T. L. Poulos, A. Pifferitti and I. F. Sevrioukova, J. Biol. Chem., 2005, 280, 16135–16142 CrossRef CAS.
- T. L. Poulos, B. C. Finzel and A. J. Howard, J. Mol. Biol., 1987, 195, 687–700 CAS.
- T. C. Pochapsky, Xiao Mei Ye, G. Ratnaswamy and T. A. Lyons, Biochemistry, 1994, 33, 6424–6432 CrossRef CAS.
- P. Wagner, FEBS Lett., 1998, 430, 112–115 CrossRef CAS.
- A. Engel and H. E. Gaub, Annu. Rev. Biochem., 2008, 77, 127–148 CrossRef CAS.
- D. J. Müller and A. Engel, Nat. Protoc., 2007, 2, 2191–2197 Search PubMed.
- V. Yu. Kuznetsov, Yu.D. Ivanov, V. A. Bykov, S. A. Saunin, I. A. Fedorov, S. V. Lemeshko, G. Hui Bon Hoa and A. I. Archakov, Proteomics, 2002, 2, 1699–1705 CrossRef CAS.
- V. Yu. Kuznetsov, Yu.D. Ivanov and A. I. Archakov, Proteomics, 2004, 4, 2390–2396 CrossRef CAS.
- O. I. Kiselyova, I. V. Yaminsky, Yu.D. Ivanov, I. P. Kanaeva, V. Yu. Kuznetsov and A. I. Archakov, Arch. Biochem. Biophys., 1999, 371, 1–7 CrossRef CAS.
- H.-J. Butt, R. Guckenberger and J. P. Rabe, Ultramicroscopy, 1992, 46, 375–393 CrossRef CAS.
- Y. Zhang, S. Sheng and Z. Shao, Biophys. J., 1996, 71, 2168–2176 CAS.
- N. H. Thomson, J. Microsc., 2005, 217, 193–199 CrossRef CAS.
- L. C. Chin, J. H. Hafner and C. M. Lieber, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3809–3813 CrossRef CAS.
- W. H. Bradshaw, H. E. Conrad, E. J. Corey, I. C. Gunsalus and D. Lednicer, J. Am. Chem. Soc., 1959, 81, 5507–5509 CrossRef CAS.
- A. I. Archakov and G. P. Bachmanova. Cytochrome P-450 and active oxygen; Taylor & Francis: London, New York, Philadelphia, 2003 Search PubMed.
- I. F. Sevriukova, H. Li and T. L. Poulos, J. Mol. Biol., 2004, 336, 889–902 CrossRef CAS.
- C. Jung, G. Hui Bon Hoa, K.-L. Schroeder, M. Simon and J. P. Doucet, Biochemistry, 1992, 31, 12855–12862 CrossRef CAS.
- I. C. Gunsalus and G. C. Wagner, Methods Enzymol., 1978, 52, 166–188 Search PubMed.
- U. Johnsson, L. Fagerstam, B. Ivarsson, B. Johnsson, R. Karlsson, K. Lundh, S. Läfas, B. Persson, H. Roos, I. Rönnberg, S. Sjölander, E. Stenberg, R. Stahlberg, C. Urbaniczky, H. Ostlin and M. Malmqvist, Bio Techniques, 1991, 11, 620–627 CAS.
| This journal is © The Royal Society of Chemistry 2010 |