Electroanalytical sensing of nitrite at shallow recessed screen printed microelectrode arrays
Mohamed
Khairy
,
Rashid O.
Kadara
and
Craig E.
Banks
*
Faculty of Science and Engineering, School of Biology, Chemistry and Health Science, Division of Chemistry and Materials, Manchester Metropolitan University, Chester Street, Manchester, M1 5GD, Lancs, UK. E-mail: c.banks@mmu.ac.uk; Fax: +44 (0)1612476831; Tel: +44 (0)1612471196
First published on 23rd April 2010
Abstract
We demonstrate that unmodified screen printed shallow recessed graphite microelectrode arrays can be used for the sensing of nitrite in aqueous solutions. The screen printed arrays comprise 6 microdiscs which have radii of 116 (±6) microns and are recessed by 4 microns and separated by 2500 microns from their nearest neighbour in a hexagonal arrangement. The screen printed arrays permit the low micromolar sensing of nitrite in aqueous solutions via cyclic voltammetry with the assessable linear range and detection limit further reduced through the application of amperometry. The protocol is shown to be feasible for the sensing of nitrite in river water samples at levels indicated by the World Health Organisation. The disposable nature and low cost of the sensor offer an economical and portable screening tool for nitrite.
Introduction
There is an unmet need for portable, reliable and economical sensors for nitrite due to its ubiquitous nature and toxicity.1,2 Nitrite is present in a plethora of samples ranging from foods as a preservative or a natural component with ingestion resulting in the formation of carcinogenic nitroso-amines in the stomach.1 In drinking water chloramination and microbial activity give rise to the formation of nitrite.3 The fatal dose of nitrite ingestion is reported as being between 8.7 µM and 28.3 µM4,5 and consequently the World Health Organisation states that nitrite should have a guidance level of 3 mg L−1 (65 µM) for short-term exposure and a provisional guideline level of 0.2 mg L−1 (4.3 µM) for long-term exposure.3 Therefore it is essential to develop reliable, portable, sensitive and economical sensors for monitoring nitrite.The range of analytical techniques has been elegantly overviewed1 with electrochemical sensors providing rapid and portable alternatives to other analytical methodologies. The electroanalytical detection of nitrite is based on either electrochemical oxidation or reduction1 with the oxidation favoured as interference from nitrate and gases present in the sample, such as oxygen, is greatly reduced. The electrochemical sensing of nitrite has been reported to be possible at a range of electrode substrates such as gold ultra-microelectrodes,5 platinum microdiscs,6 glassy carbon7,8 and boron-doped diamond.9 Problems can occur in some instances when using bare electrode substrates that cumulative electrode passivation/fouling effects can occur decreasing the sensitivity of the technique.1 Due to these reported problems, research is currently directed towards the use of electrodes modified with electro-catalytic compounds.
Compton and co-workers have re-visited the electrochemical oxidation of nitrite at bare, unmodified glassy carbon electrodes arguing that the modification of electrode substrates with electro-catalytic materials only serves to shift the voltammetric peak potential to less oxidative potentials. Associated with this is a negligible (∼1.3) increase in the magnitude of the voltammetric peak, which is unlikely, in the case of nitrite, to have a major benefit to the analytical performance of the sensor.8 Compton et al. went on to demonstrate that passivation effects, that are encountered at the bare unmodified glassy carbon electrode, could be overcome by employing the use of power ultrasound to clean the electrochemical surface.8 This technique is very promising as a semi-portable methodology for the sensing of nitrite. Another approach would be to employ the use of screen printed electrodes which can act voltammetrically similar to solid carbon electrode substrates but are extremely attractive due to their low cost and disposable nature. Despite their advantages, bare screen printed electrodes have not been extensively explored and the work of Kalcher et al.10 describing screen printed electrodes fabricated using carbon ink modified with an anion-exchanger and screen printed edge band ultra-microelectrodes have only been reported.11
Recently we have reported the fabrication of shallow recessed graphite microelectrode arrays fabricated purely by screen printing for the sensing of manganese(II)12 and lead(II) in river water.13 Microelectrode arrays are useful electrode platforms in the electrochemists' arsenal allowing lower detection limits and greater sensitivities to be achieved in comparison to macroelectrodes.14–19 To date there are no reports of using a microelectrode array for the sensing of nitrite. We report for the first time the electroanalytical sensing of nitrite at a disposable screen printed shallow recessed microelectrode array which allows the low micromolar sensing of nitrite in river water samples at levels suggested by the World Health Organisation.
Experimental section
All chemicals used were of analytical grade and were used as received without any further purification and were obtained from Sigma-Aldrich. All solutions were prepared with deionised water of resistivity not less than 18.2 MΩ cm−1.Voltammetric measurements were carried out using a µ-Autolab III (ECO-Chemie, The Netherlands) potentiostat. The microelectrode arrays were fabricated in-house using a DEK 1760RS screen-printing machine (DEK, Weymouth, UK). A carbon–graphite ink (Gwent Electronic Materials Ltd, Pontypool, UK) was first screen printed onto a polyester flexible film (Autostat AHU10, 250 µm thickness), supplied by Autotype (Oxon, UK). The layer was cured in a fan oven at 60 degrees for 30 minutes. Onto this, a dielectric paste ink (Gwent Electronic Materials Ltd, Pontypool, UK) was printed which is used to define the working microelectrodes array. In this case, microsized holes in the screen are blocked thus leaving the underlying carbon–graphite layer exposed. An onboard reference electrode was included by printing Ag/AgCl paste ink (Gwent Electronic Materials Ltd, Pontypool, UK) onto the initially printed carbon–graphite track. The screen printed shallow recessed graphite microelectrode arrays comprise 6 microdiscs which have radii of 116 (±6) microns which are recessed by 4 microns and are separated by 2500 microns from their nearest neighbour in a hexagonal arrangement. The screen printed microelectrode arrays are produced with an onboard carbon counter and Ag/AgCl reference electrode. However, in most cases a Saturated Calomel Electrode (SCE) was used for comparative purposes. Connectors for the efficient connection of the screen printed electrochemical sensors were purchased from Kanichi Research Services Ltd (UK).20 Solutions in the range pH 1 to pH 12 were prepared as follows: pH 1 to pH 3: hydrochloric acid, pH 4.6: 0.1 M acetic acid and 0.1 M sodium acetate, pH 6.8: sodium phosphate dibasic dihydrate and sodium phosphate monobasic dihydrate, pH 9.2: disodium tetraborate, pH 10 to pH 12: sodium hydroxide. All solutions contained 0.1 M potassium chloride as supporting electrolyte. River water was sampled from ‘Jacksons Boat’ bridge over River Mersey, Sale, Cheshire, UK and was simply acidified to the appropriate pH with small amounts of concentrated hydrochloric acid.
Results and discussion
The voltammetric response towards the electrochemical oxidation of nitrite was explored using shallow recessed screen printed microelectrode arrays in a 1 mM nitrite solution in a pH 3 buffer solution where as shown in Fig. 1 a large voltammetric profile is observed. Additionally shown is the response of the sensor towards nitrate which clearly indicates that nitrate is electrochemically inactive in the explored potential window indicating specificity towards nitrite which is extremely important given that nitrite and nitrate are usually simultaneously present in samples. Previous results at bare electrode substrates have shown that the electrochemical oxidation of nitrite results in cumulative electrode passivation/fouling effects which greatly limits the analytical performance1 of the sensor and is a key reason as to why chemically modified electrodes have been developed. In a 1 mM nitrite solution the effect of continuous voltammetric scans was explored and the first and tenth voltammetric scans are depicted in Fig. 1A where no noticeable reduction in the voltammetric performance is observed indicating that the screen printed microelectrode array has potential as a nitrite sensor.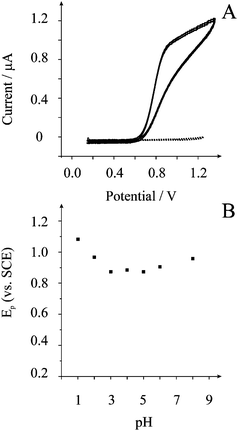 | ||
| Fig. 1 Typical cyclic voltammograms (A) of the screen printed microelectrode array in 1 mM nitrate (dotted line) and 1 mM nitrite (solid line) in a pH 3 solution. Scan rate: 0.1 V s−1vs. SCE. The response is also shown for the 10th continuous voltammetric scan in 1 mM nitrite (solid line). Peak potential dependence on solution pH (B). | ||
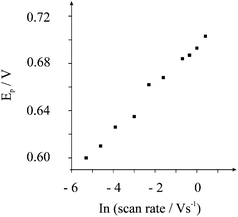
The voltammetric response towards the electrochemical oxidation of nitrite was explored using shallow recessed screen printed microelectrode arrays in nitrite solutions of differing pHs. Analysis of the voltammetric peak potential, Ep, at each pH is shown in Fig. 1B. These parameters were extracted at the point in the voltammetric profile corresponding just before the onset of the steady-state portion of the profile. It is evident that a linear response of peak potential from the oxidation of nitrite with pH is observed. Beyond pH 3 the peak potential is insensitive to pH which is consistent with the reported pKa for nitrous acid being between 3.2 and 3.4.21,22 At low pH values the electrochemical signal is positioned sufficiently away from the potential window which ends at approx. +1.3 V. Cyclic voltammetric experiments were performed in pH 3 over the range of scan rates from 0.005 to 1.0 V s−1 where the observed voltammetric profiles are gentle peaks rather than steady-state currents which is clearly evident from the voltammetry presented in Fig. 1. A signal–scan rate dependence for all of the scan rates was observed—upon increasing the scan rate, the magnitude of the I–E response increases where the peak current is proportional to the square root of scan rate. The lack of true steady state behaviour indicates a degree of diffusion interaction between neighbouring microdiscs. Analysis of the limiting currents using the Tomeš criterion (E3/4 − E1/4), which is the difference between the three-quarter wave potential and quarter wave potential, found this to be 95 mV suggesting an electrochemically irreversible system.23 For a totally irreversible diffusion-controlled processes, as is expected in light of previous studies,8,21,24 the analysis of the peak potential against scan rate, as depicted in Fig. 2, allows mechanistic information to be obtained from eqn (1):
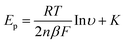 | (1) |
| NO−2 +H+ ⇌ HNO2 | (2) |
| NO−2 − e ⇌ NO2 | (3) |
| 2NO2 +H2O → NO−3 +NO−2 +2H+ | (4) |
 | ||
| Fig. 2 Variation of peak potential, Ep, as a function of scan rates performed at pH 3 using the screen printed microelectrode array. | ||
Next attention was turned to exploring the electroanalytical performance of the screen printed microelectrode array towards nitrite detection. Additions of nitrate were made into a pH 3 solution over the range 20 µM to 400 µM. Fig. 3 depicts the voltammetric responses resulting from the additions of nitrite and analysis of the peak height versus nitrite concentration is also shown where a linear response is observed over the range 20 µM to 340 µM (Ip/A = 6.6 × 10−4 A/M + 8.3 × 10−9 A; R2 = 0.997; N = 17). Based on this linear range, the limit of detection (3σ)27 was found to correspond to 12.7 µM. This analytical range is suitable for determining short-term exposure as indicated by the World Health Organisation.3 Additionally we note that this limit of detection is less than that obtainable at a glassy carbon macroelectrode8 and similar to that previously reported using gold ultramicroelectrodes,5 carbon fibre microelectrodes,26 and platinum microdisc electrodes using amperometry.6 The reason for this lies in the fact that in the use of microelectrodes, over previously used macroelectrodes, radial diffusion dominates which clearly has beneficial analytical utility compared to conventional carbon electrodes.
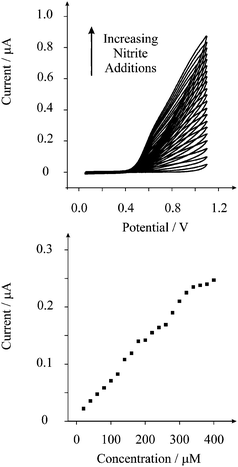 | ||
| Fig. 3 Typical cyclic voltammograms of nitrite into a pH 3 solution using the screen printed microelectrode array. Also shown is the analysis of the current as a function of nitrite concentration. | ||
To access the electroanalytical performance of the screen printed microelectrode array we turned to exploring the sensing of nitrite in river water. Note that the only pre-treatment was the addition of a small amount of acid to alter the pH of the sample to 3. The voltammetric performance of the screen printed microelectrode array towards nitrite detection in the river water sample was explored but no detectable nitrite was found indicating that the nitrite occurs at a low level, as expected, which is below our detection limit capabilities. Consequently a recovery experiment was performed on the river water sample. The sample was spiked with 20 µM of nitrite with a standard addition protocol performed in triplicate; Fig. 4 depicts a typical plot. Based on these recovery experiments, it was found that a recovery of 88 (±1)% was possible.
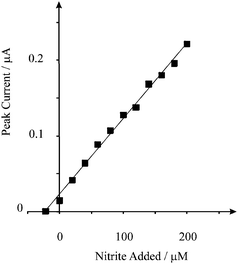 | ||
| Fig. 4 Standard additions of nitrite into a river water sample using the screen printed microelectrode array. The sample was simply acidified to pH 3 with a small amount of concentrated hydrochloric acid. | ||
Next the amperometric response of the screen printed microelectrode arrays was explored in order to increase the sensitivity of the protocol. Fig. 5 depicts a typical amperometric response where analysis as a function of concentration reveals a linear response over the range 1 µM to 17 µM (Ip/A = 1.4 × 10−3 A/M + 2.3 × 10−9 A; R2 = 0.999; N = 18) and based on this linear range, the limit of detection (3σ)27 was found to correspond to 0.3 µM. This limit of detection is comparable to screen-printed edge band ultramicroelectrodes11 and that of many modified electrode substrates.8 Additionally the accessible linear range and detection limit are suitable for the determination of long-term exposure as governed by the World Health Organisation.3 The disposable shallow recessed graphite microelectrode arrays comprise microelectrodes which have radii of 116 and, in terms of other available microelectrode arrays, are relatively large. We expect that if arrays with smaller radii are used but yet with sufficient distance between neighbouring microelectrodes, further enhancements in the analytical performance will likely be achieved. However, the benefits of a disposable and economical array based on screen printing are clearly evident.
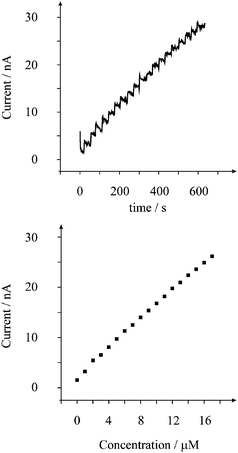 | ||
| Fig. 5 Amperometric response (top) from the additions of nitrite into pH 3 solution using the screen printed microelectrode array. The current was held at +0.75 V. Also shown is the analysis of the amperometric response (bottom) as a function of nitrite concentration. | ||
Conclusions
We have demonstrated that screen printed microelectrode arrays can be used for the low micromolar sensing of nitrite without recourse to chemical modification or power ultrasound.8 The sensing of nitrite is shown to be possible in river water samples and the detection limit and linear range can be further reduced by the application of amperometry. Given the disposable nature and low cost of the screen printed microelectrode arrays the portable sensing of nitrite is extremely promising.References
- M. J. Moorcroft, J. Davis and R. G. Compton, Talanta, 2001, 54, 785 CrossRef CAS.
- G. Ellis, I. Adatia, M. Yazdanpanah and S. K. Makela, Clin. Biochem., 1998, 31, 195 CrossRef CAS.
- http://www.who.int/water_sanitation_health/dwq/gdwq3_8.pdf .
- Drinking Water Standards—1962, US Department of Health, Education and Welfare, Public Health Service, Washington, DC, 1962, p. 47 Search PubMed.
- S. M. Silva and L. Henrique Mazo, Electroanalysis, 1998, 10, 1200 CrossRef CAS.
- M. Bertotti and D. Pletcher, J. Braz. Chem. Soc., 1997, 8, 391 CAS.
- A. Y. Chamsi and A. G. Fogg, Analyst, 1988, 113, 1723 RSC.
- B. R. Kozub, N. V. Rees and R. G. Compton, Sens. Actuators, B, 2010, 143, 539 CrossRef.
- N. Spataru, T. N. Rao, D. A. Tryk and A. Fujisjima, J. Electrochem. Soc., 2001, 148, E112 CrossRef CAS.
- C. G. Neuhold, J. Wang, X. H. Cai and K. Kalcher, Analyst, 1995, 120, 2377 RSC.
- J.-L. Chang and J.-M. Zen, Electroanalysis, 2006, 18, 941 CrossRef CAS.
- R. O. Kadara, N. Jenkinson and C. E. Banks, Sens. Actuators, B, 2009, 142, 342 CrossRef.
- S. J. Hood, R. O. Kadara, D. K. Kampouris and C. E. Banks, Analyst, 2010, 135, 76 RSC.
- S. Ward-Jones, C. E. Banks, A. O. Simm, L. Jiang and R. G. Compton, Electroanalysis, 2005, 17, 1806 CrossRef CAS.
- T. J. Davies, S. Ward-Jones, J. del Campo, R. Mas, F. X. Munoz and R. G. Compton, J. Electroanal. Chem., 2005, 585, 51 CrossRef CAS.
- A. O. Simm, C. E. Banks, S. Ward-Jones, T. J. Davies, N. S. Lawrence, T. G. J. Jones, L. Jiang and R. G. Compton, Analyst, 2005, 130, 1303 RSC.
- O. Ordeig, J. del Campo, F. X. Munoz, C. E. Banks and R. G. Compton, Electroanalysis, 2007, 19, 1973 CrossRef CAS.
- V. Beni and D. V. M. Arrigan, Curr. Anal. Chem., 2008, 4, 229 CrossRef CAS.
- S. Fletcher and M. D. Horne, Electrochem. Commun., 1999, 1, 502 CrossRef CAS.
- http://kanichi-research.com/ .
- X. Huang, Y. Li, Y. Chen and L. Wang, Sens. Actuators, B, 2008, 134, 780 CrossRef.
- G. Milczarek, Electroanalysis, 2008, 20, 211 CrossRef CAS.
- Y. Zhang, C. D. Baer, C. Camaioni-Neto, P. O'Brien and D. A. Sweigart, Inorg. Chem., 1991, 30, 1682 CrossRef CAS.
- R. Guidelli, F. Pergola and G. Raspi, Anal. Chem., 1972, 44, 745 CrossRef CAS.
- X. Xing and D. A. Scherson, Anal. Chem., 1988, 60, 1468 CrossRef CAS.
- S. M. Silvu, C. R. Alves, S. A. S. Muchudo, L. H. Muzo and L. A. Avucu, Electroanalysis, 1996, 8, 1055.
- C. M. A. Brett and A. M. O. Brett, Electroanalysis, Oxford University Press, Oxford, 1998 Search PubMed.
| This journal is © The Royal Society of Chemistry 2010 |