Electrochemical determination of oxalic acid using palladium nanoparticle-loaded carbon nanofiber modified electrode
Yang
Liu
ab,
Jianshe
Huang
ab,
Dawei
Wang
ab,
Haoqing
Hou
*c and
Tianyan
You
*a
aState Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, China. E-mail: youty@ciac.jl.cn; Fax: +86-431-85262850; Tel: +86-431-85262850
bGraduate School of the Chinese Academy of Sciences, Beijing, 100039, China
cCollege of Chemistry and Chemical Engineering, Jiangxi Normal University, Nanchang, 330027, China
First published on 19th May 2010
Abstract
We have developed a novel electrochemical oxalic acid (OA) sensor based on the palladium nanoparticle-loaded carbon nanofiber (Pd/CNF) composites. These composites with large amounts of spherical nanoparticles well dispersed on the carbon nanofibers (CNF) were produced by combination of electrospinning technique with thermal treatment method. When applied to oxidation of OA, the Pd/CNF modified carbon paste electrode (Pd/CNF-CPE) exhibited high electrocatalytic performances with fast voltammetric responses and notably decreased overpotential compared to the bare and even the CNF modified CPE. A detection limit of 0.2 mM with linear ranges of 0.2–13 mM and 13–45 mM can be obtained at the Pd/CNF-CPE. Based on its high sensitivity and good selectivity, the proposed method was applied to determination of OA in spinach, and the satisfactory results confirmed the applicability of this sensor in practical analysis.
1. Introduction
Oxalic acid (OA) widely exists in plants, animals and microbes. Since it can easily combine with Ca2+ and Mg2+ to form less soluble salts, high levels of OA in the digestive system lead to formation of kidney stones. Therefore, the accurate detection of OA has attracted considerable interest in arrangement of a rational diet and management of food qualities, as well as in diagnosis and prevention of renal stone formation. Various approaches have been proposed to meet the rising demands for OA sensors, such as spectrophotometry,1,2 gas/liquid chromatography,3,4 and enzymatic methods.5,6 However, the practical applications of these methods are limited by relatively high cost and delicate procedures compared to electrochemical techniques. In this regard, novel electrode materials are highly desirable for the development of electrochemical OA sensors with high sensitivity and good selectivity.Carbon electrodes have been widely used in electrochemistry due to their low cost, wide potential window and chemical inertness.7 Several carbon materials have been investigated for electrochemical oxidation of OA, such as boron-doped diamond,8 and acid-pretreated carbon nanotubes.9 Although low detection limit and good stability were obtained at these electrodes, OA oxidation required high overpotential and the electrochemical performances were highly dependent on the pretreatment of the carbon materials.10 Many other modified electrodes were proposed to lower the overpotential for OA oxidation, such as the exfoliated graphite-polystyrene composite electrode,11 and the Ti/PbO2 electrode.12 With the rapid progress in nanotechnology, palladium nanoparticles (Pd NPs) have become one of the most frequent choices for electrode modification because of their high catalytic activities towards many electrochemical reaction,13–15 and relatively low cost compared to Pt NPs. Based on their attractive properties, Pd NPs were deposited on the traditional electrodes for electrocatalytic oxidation of OA with high response and low overpotential.16–18 Nowadays, novel Pd NP-based materials with good dispersity and high catalytic efficiency are still attractive for practical analysis of OA.
In this work, a novel electrochemical OA sensor was proposed based on electrospun palladium nanoparticle-loaded carbon nanofibers modified carbon paste electrode (Pd/CNF-CPE). This electrode exhibited attractive performances for OA detection, such as strong and fast voltammetric response, as well as good selectivity in the presence of ascorbic acid (AA) and uric acid (UA). The sensor was applied to a real matrix; OA in spinach was successfully detected at the Pd/CNF-CPE with satisfactory results.
2. Experimental
2.1. Apparatus
The scanning electron microscopy (SEM) experiment was made on a PHILIPS XL-30 ESEM at an accelerating voltage of 20 kV. The transmission electron microscope (TEM) images were obtained using a JEOL 2000 TEM (JEOL, Japan) operating at 200 kV. The X-ray powder diffraction (XRD) analysis was carried out on a Bruker D8 ADVANCE instrument with Cu-Kα1 radiation (40 kV, 40 mA). All electrochemical experiments were performed with a CHI832 electrochemical workstation (China). A conventional three-electrode system was used, which consisted of a platinum wire as auxiliary electrode, a Ag/AgCl/saturated KCl as reference electrode, and a bare or modified CPE as working electrode.2.2. Chemicals and reagents
Polyacrylonitrile (PAN), Pd(acetate)2 (Pd(Ac)2), and dimethylformamide (DMF) were purchased from Aldrich. Oxalic acid (OA), ascorbic acid (AA), uric acid (UA) and HClO4 were obtained from Beijing Chemical Reagents Company (China). All aqueous solutions were prepared with double distilled water.2.3. Preparation of spinach sample
The spinach samples were prepared as previously described.19 Briefly, 2 g of spinach was cut into small pieces using a razor blade, and was boiled with water under reflux for 50 min. The mixed suspension was centrifuged at 3000 rpm for 3 min. The supernatant was filtered through 0.45 μm filter paper, and then diluted to 10 ml in a calibrated flask.2.4. Preparation of Pd/CNF composites
The Pd/CNF composites were prepared by carbonizing the electrospun PAN/Pd(Ac)2 composite fibers, which were made via electrospinning process using a DMF solution containing 8 wt% PAN and 4.8 wt% Pd(Ac)2. The detail process was similar to the previous reports.20,21 Briefly, the electrospinning process was performed in the electric fields of the order of 100 kV m−1, from a 30 kV voltage applied to a 30 cm gap between the spinneret and the collector. Stabilization and carbonization of PAN, and the reduction of Pd2+ were completed in a high-temperature furnace by the following steps: (1) 230 °C annealing in air for 3 h to oxidize partially the polymer PAN; (2) heating up to 300 °C at a rate of 5 °C min−1 and stayed at this temperature in H2 and Ar mixture (H2/Ar = 1/3) for 2 h for the shape stabilization of the nanofibers and the reduction of Pd2+; (3) heating up to 1100 °C in Ar at a rate of 5 °C min−1 to carbonize the nanofibers, staying at the highest temperature for half an hour, and then cooling down to room temperature in Ar.2.5. Preparation of electrodes
Carbon paste electrode (CPE) was prepared by mixing graphite powder and mineral oil at the ratio of 70/30 (w/w). The paste was carefully hand-mixed in a mortar and then packed into a pipette tube (1.2-mm diameter; 1-cm depth). The electrical contact was provided by a copper wire connected to the paste in the inner hole of the tube. Pd/CNF-CPE was prepared by casting 1 mg mL−1 Pd/CNF (10 μL) suspension on the surface of CPE and allowed to dry in a desiccator at room temperature. The resulting Pd/CNF-CPE was rinsed carefully with double-distilled water prior to each measurement. When the modified electrode was not in use, it was stored in a desiccator at room temperature.3. Results and discussion
3.1. Characterization of Pd/CNF composites
Great efforts have been made for preparation of Pd/CNF composites, which combine the advantages of high catalytic efficiency, good electronic properties and unique mechanical strength.22–24 Regarding the fabrication procedures, two separate steps are necessary, namely, synthesis of the CNF supports and deposition of the Pd catalysts. However, metal nucleation requires modification of the CNF surface, which makes the procedure tedious and prone to introduce impurities. In this work, PAN and Pd(Ac)2 were used as the precursors of CNF and Pd NPs, respectively. During the conversion of electrospun PAN nanofibers to CNF in a high temperature furnace, Pd2+ in the polymer nanofibers can be reduced to Pd0 and aggregated into Pd NPs simultaneously. It means that the Pd/CNF composites are formed in one-step procedure, which may significantly simplify the preparation process as well as improve the performances of the composite materials.TEM was used to investigate the morphology of the Pd/CNF composites. As shown in Fig. 1a, the diameter of the CNF ranges from 300 to 500 nm with tens of micrometre in length, and large amounts of nanoparticles are well dispersed on the surface of the CNF. The high-magnification image illustrates that the nanoparticles have a diameter of about 100 nm and are firmly embedded in the CNF matrix (Fig. 1b).
 | ||
| Fig. 1 TEM images of Pd/CNF composites at (a) low and (b) high magnifications. | ||
The XRD pattern demonstrates the constitution of the Pd/CNF composites (Fig. 2). It shows five characteristic diffraction peaks at 40.4°, 46.9°, 68.6°, 82.6° and 87.1, corresponding to the (111), (200), (220), (311) and (222) crystalline planes of cubic Pd, respectively (JCPDS, No. 65-2867). Herein, the graphitic structure of CNF was not observed, which is in agreement with the previous report that the PAN-derived CNF are non-graphitizable carbon, even at a thermal treatment temperature of 2800 °C.25
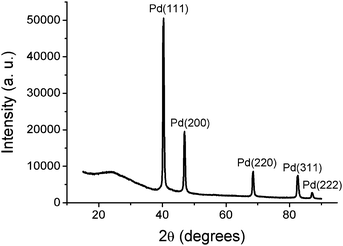 | ||
| Fig. 2 XRD pattern of Pd/CNF composites. | ||
3.2. Characterization of Pd/CNF-CPE
The Pd/CNF composites were modified at the surface of the CPE to investigate their electrocatalytic performances. Firstly, the surface morphologies of the bare CPE, CNF modified CPE (CNF-CPE) and the Pd/CNF modified CPE (Pd/CNF-CPE) were characterized by SEM for comparison (Fig. 3). As shown in Fig. 3a, the surface of CPE is formed by irregularly lumpy and flaky graphite in micrometre size. After modification with CNF, the electrode surface was covered by a network of plentiful smooth nanofibers (Fig. 3b). In contrast, large amounts of Pd NPs with good dispersivity were observed at the Pd/CNF-CPE, which indicates that the composites can be successfully modified on the surface of the electrode (Fig. 3c). | ||
| Fig. 3 SEM images of surface morphology of (a) bare CPE, (b) CNF-CPE and (c) Pd/CNF-CPE. | ||
In addition, the electrochemical behavior of the Pd/CNF-CPE in acid medium is similar to that of other Pd-based electrodes.17,26,27 During the positive potential scan, the metallic Pd0 was oxidized to Pd oxides when the applied potential increased to about 0.6 V. After that, the oxygen evolution was accompanied with the formation of higher oxidation state of Pd when the potential was more than 1.2 V.28 On the subsequent negative sweep, the corresponding oxides were reduced with the applied potential decreased, and a clear surface of Pd0 could be reformed on the electrode.
3.3. Electrocatalytic oxidation of OA
To investigate the electrocatalytic activity of the Pd/CNF-CPE towards OA oxidation, control cyclic voltammetry experiments were carried out at the bare CPE, CNF-CPE and Pd/CNF-CPE in 0.1 M HClO4 containing 10 mM OA (Fig. 4). As illustrated, the oxidation of OA at the CPE shows a broad peak at 1.55 V (solid line), and similar electrochemical behavior can be observed at the CNF/CPE (dot line). Interestingly, when the Pd/CNF-CPE was used, a fast voltammetric response to OA oxidation is obtained at 1.10 V, indicating a remarkable negative shift by more than 0.40 V (dash line). It means that the Pd NPs dispersed on the electrode surface have significant effect on the electrocatalytic oxidation of OA.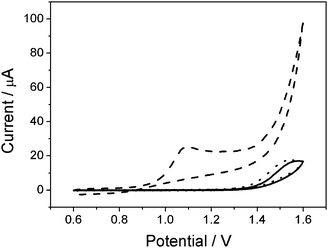 | ||
| Fig. 4 CVs of CPE (solid), CNF-CPE (dot) and Pd/CNF-CPE (dash) in 0.1 M HClO4 containing 10 mM OA. Scan rate: 0.05 V s−1. | ||
Regarding the electrochemical behavior of OA oxidation, various electrode materials have been proposed for investigation, such as Pt,29,30 Au,31,32 Pd,17,31 and WOx.33 As De Battisti et al. pointed out, the reaction mechanism is highly dependent on the nature of electrode material. The oxidation reaction is fast when the interaction of OA molecules with electrode surface is particularly strong.12 Vasil'ev and Sargisyan revealed that the oxidation of OA is completely inhibited at the glassy carbon electrode because of a scarce adsorptive ability. And its rate determined on some other electrode materials decreases as follows: Pd ≈ Au ≥ Pt > Ir > Rh.30 In this regard, OA molecules tend to adsorb on the Pd0 sites of the Pd/CNF-CPE as follows:
| Pd0 + H2C2O4 → Pd(H2C2O4)ads |
| Pd0 → Pd2+ + 2e− |
Meanwhile, the adsorbed complex undergoes a probable fast concerted hydrogen abstraction mechanism according to the following reaction:
| (H2C2O4)ads + Pd2+ → Pd + 2CO2 + 2H+ |
As illustrated, the oxidation of OA at the Pd/CNF-CPE is associated with the formation of the Pd ion, which is in agreement with the previous report,17 so that the oxidation potential greatly decreased compared to that observed at the bare CPE and even the CNF/CPE (Fig. 4).
3.4. Determination of OA by DPV at Pd/CNF-CPE
Fig. 5a demonstrates the DPV responses of the Pd/CNF-CPE towards different concentrations of OA. A detection limit of 0.2 mM (S/N = 3) can be achieved, and the calibration curve for OA detection are linear in two ranges of 0.2–13 mM and 13–45 mM (Fig. 5b), which is wider than that obtained at the exfoliated graphite-polystyrene composite electrode,11 and comparable to those obtained at the boron-doped diamond electrode8 as well as the carbon nanotubes modified electrode.9 Additionally, the Pd/CNF-CPE notably decreased the overpotential for OA oxidation compared to those carbon based electrodes, attributing to the high electrocatalytic performances of the Pd NPs.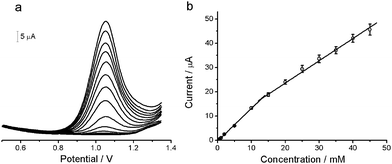 | ||
| Fig. 5 (a) DPVs at Pd/CNF-CPE in 0.1 M HClO4 containing different concentrations of OA (from inner to outer): 0.2, 0.4, 0.6, 1, 2, 5, 10, 15, 20, 25, 30, 35, 40, 45 mM. (b) The calibration curve for OA detection. DPV conditions: amplitude, 50 mV; pulse width, 50 ms; pulse period, 200 ms. | ||
Another attractive feature of the Pd/CNF-CPE is the good selectivity for detection of OA in the presence of AA and UA, which are common electroactive interferents in practical analysis. Fig. 6 shows the DPV responses to the mixture of OA, AA and UA at the Pd/CNF-CPE. Three well-defined peaks are observed at 0.37, 0.68 and 1.07 V, corresponding to the oxidation of AA, UA and OA, respectively. The peak separations of these three species are large enough for simultaneous determination, so that OA can be selectively detected at the Pd/CNF-CPE without interference from AA and UA.
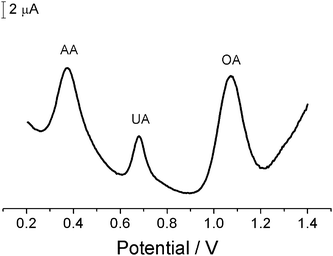 | ||
| Fig. 6 DPV at Pd/CNF-CPE in 0.1 M HClO4 containing 10 mM AA, 0.5 mM UA and 10 mM OA. DPV conditions as in Fig. 5. | ||
3.5. Reproducibility and stability of Pd/CNF-CPE
Besides sensitivity and selectivity, reproducibility and stability are important parameters to evaluate the applicability of electrochemical sensors. Herein, the batch-to-batch reproducibility of the Pd/CNF-CPE was first estimated from the current responses of six different electrodes to 5 mM OA. The relative standard deviation (RSD) was calculated to be 4.8%, which is acceptable for sensor fabrication. In addition, the Pd/CNF-CPE showed RSD of 3.8% examined for five successive measurements of 3 mM OA. When the sensor was stored in a desiccator at room temperature for one month, the current response remained 96% of its initial value, suggesting the good stability of the electrode. Therefore, the Pd/CNF-CPE is promising for determination of OA with acceptable reproducibility and long-term stability.3.6. Sample analysis
In order to verify the reliability of the proposed method, the Pd/CNF-CPE was applied to analysis of OA in spinach sample. The sample extraction was diluted 5 times with 0.1 M HClO4 before transferred to an electrochemical cell for DPV measurement. The analytical results are summarized in Table 1. As indicated, the RSD is less than 3.1% and the recovery ranges from 97.7% to 105.3%, which can be acceptable for practical analysis. In addition, based on the same procedures for sample preparation, the content of OA detected using the proposed method was 4.29 ± 0.13 mg g−1, close to 4.15 ± 0.16 mg g−1 reported in the literature.19 Therefore, the satisfactory results obtained with this method confirm the strong applicability of the Pd/CNF-CPE in practical analysis.4. Conclusions
In summary, a novel electrochemical OA sensor was presented using the Pd/CNF composites, which were produced by combination of electropinning technique and thermal treatment method. In application to electrocatalytic oxidation of OA, the Pd/CNF-CPE embedded with large amounts of Pd NPs, exhibited strong and fast voltammetric response with low overpotential. The attractive analytical performances obtained with the proposed OA sensor such as the wide linear range, good selectivity and strong applicability to real samples, may potentially pave the way for management of food qualities in markets and diagnosis of renal stone formation in clinical medicine.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (No. 20875085) and Chinese Academy of Sciences (KJCX2-YW-H11).References
- M. F. Laker, A. F. Hofman and B. J. D. Meeuse, Clin. Chem., 1980, 26, 827–830 CAS.
- A. A. Ensafi and A. Kazemzadeh, Fresenius J. Anal. Chem., 2000, 367, 590–592 CrossRef CAS.
- E. J. Jellum, J. Chromatogr., B: Biomed. Sci. Appl., 1977, 143, 427–462 CrossRef CAS.
- F. W. Wu, Z. K. He, Q. Y. Luo and Y. E. Zeng, Food Chem., 1999, 65, 543–546 CrossRef CAS.
- S. M. Reddy, S. P. Higson and P. M. Vadgama, Anal. Chim. Acta, 1997, 343, 59–68 CrossRef CAS.
- E. F. Perez, G. De Oliveira Neto and L. T. Kubota, Sens. Actuators, B, 2001, 72, 80–85 CrossRef.
- R. L. McCreery, Chem. Rev., 2008, 108, 2646–2687 CrossRef CAS.
- T. A. Ivandini, T. N. Rao, A. Fujishima and Y. Einaga, Anal. Chem., 2006, 78, 3467–3471 CrossRef CAS.
- Y. Zheng, C. Yang, W. Pu and J. Zhang, Food Chem., 2009, 114, 1523–1528 CrossRef CAS.
- B. Šljukić, R. Baron and R. G. Compton, Electroanalysis, 2007, 19, 918–922 CrossRef CAS.
- F. Manea, C. Radovan, I. Corb, A. Pop, G. Burtica, P. Malchev, S. Picken and J. Schoonman, Sensors, 2007, 7, 615–627 CrossRef CAS.
- C. A. Martínez-Huitle, S. Ferro and A. D. Battisti, Electrochim. Acta, 2004, 49, 4027–4034 CrossRef CAS.
- J. Wang, N. Naser, L. Angnes, H. Wu and L. Chen, Anal. Chem., 1992, 64, 1285–1288 CrossRef CAS.
- J. Liu, W. H. Zhou, T. Y. You, F. L. Li, E. K. Wang and S. J. Dong, Anal. Chem., 1996, 68, 3350–3353 CrossRef CAS.
- D. J. Guo and H. L. Li, Electrochem. Commun., 2004, 6, 999–1003 CrossRef CAS.
- L. G. Shaidarova, A. V. Gedmina, I. A. Chelnokova and G. K. Budnikov, J. Anal. Chem., 2003, 58, 886–891 CrossRef CAS.
- I. G. Casella, Electrochim. Acta, 1999, 44, 3353–3360 CrossRef CAS.
- L. G. Shaidarova, I. A. Chelnokova, A. V. Gedmina, G. K. Budnikov, S. A. Ziganshina, A. A. Mozhanova and A. A. Bukharaev, J. Anal. Chem., 2006, 61, 375–381 CrossRef CAS.
- A. Safavi and A. R. Banazadeh, Food Chem., 2007, 105, 1106–1111 CrossRef CAS.
- J. Huang, D. Wang, H. Hou and T. You, Adv. Funct. Mater., 2008, 18, 441–448 CrossRef CAS.
- J. Huang, Y. Liu, H. Hou and T. You, Biosens. Bioelectron., 2008, 24, 632–637 CrossRef CAS.
- P. Serp, M. Corrias and P. Kalck, Appl. Catal., A, 2003, 253, 337–358 CrossRef CAS.
- W. Q. Yang, S. H. Yang, J. S. Guo, G. Q. Sun and Q. Xin, Carbon, 2007, 45, 397–401 CrossRef CAS.
- J. S. Zheng, X. S. Zhang, P. Li, J. Zhu, X. G. Zhou and W. K. Yuan, Electrochem. Commun., 2007, 9, 895–900 CrossRef CAS.
- C. Kim, K. S. Yang, M. Kojima, K. Yoshida, Y. J. Kim, Y. A. Kim and M. Endo, Adv. Funct. Mater., 2006, 16, 2393–2397 CrossRef CAS.
- L. D. Burke and J. K. Casey, J. Electrochem. Soc., 1993, 140, 1284–1291 CAS.
- L. D. Burke and L. C. Nagle, J. Electroanal. Chem., 1999, 461, 52–64 CrossRef CAS.
- K. Gossner and E. Mizera, J. Electroanal. Chem., 1981, 125, 347–358 CAS.
- S. A. Sargisyan and Yu. B. Vasil'ev, Soviet Electrochem., 1982, 18, 848–853.
- S. A. Sargisyan and Yu. B. Vasil'ev, Soviet Electrochem., 1982, 18, 854–857.
- N. A. Morozova, G. E. Shcheblykina and A. V. Vvedenskii, Russ. J. Electrochem., 1999, 35, 310–319 CAS.
- R. Albalat, E. Gomez, M. Sarret and E. Valles, Monatsh. Chem., 1989, 120, 651–659 CrossRef CAS.
- C. Bock, A. Smith and B. MacDougall, Electrochim. Acta, 2002, 48, 57–67 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |