Novel extraction method based on the dispersion of the extraction solvent for extraction of letrozole from biological fluids
Mohammad
Rezaee
,
Yadollah
Yamini
*,
Mohammad
Hojjati
and
Mohammad
Faraji
Department of Chemistry, Faculty of Sciences, Tarbiat Modares University, P.O. Box 14115-75, Tehran, Iran. E-mail: yyamini@modares.ac.ir.; Fax: +98-21-88006544; Tel: +98-21-82883417
First published on 2nd August 2010
Abstract
The analysis of drugs in various biological fluids is an important criterion for the determination of the physiological performance of a drug. Dispersive liquid–liquid microextraction (DLLME) technique was successfully used as a simple, rapid and sensitive sample preparation method for the determination of letrozole in biological fluids and water samples. Chloroform at microlitre volume level and acetone were used as extraction and disperser solvents, respectively. The influence of several variables (e.g. type and volume of disperser and extraction solvents, ionic strength, etc.) on the performance of the sample preparation step was carefully evaluated. Under the optimum conditions, a preconcentration factor of 120 was obtained from only 10.0 mL of a water sample. The calibration graph was linear in the range of 1–500 μg L−1 with a detection limit of 0.3 μg L−1. The relative standard deviation (RSD) for five replicate measurements of letrozole was 3.2%. The calibration graphs were linear in the range of 2.5–500 μg L−1 and 5.0–500 μg L−1 with detection limits of 0.7 μg L−1 and 1.5 μg L−1 in urine and plasma samples, respectively. The relative recoveries of letrozole in plasma, urine and tap water samples at spiking levels of 50 μg L−1, 30 μg L−1 and 5 μg L−1 were 87.4%, 92.0% and 96.0%, respectively. DLLME combined with HPLC-UV is a fast, simple and efficient method for the determination of letrozole in biological fluids and water samples.
1. Introduction
Today with new drugs we are finding the need to know the concentration of drugs within their subject, then later what happens when they are expelled from the body into the ecosystem. One way is by monitoring the concentration of the drugs in various biological fluids. Drug analysis in biological systems provides valuable information about potential bioavailability, metabolism, toxicology, pharmacokinetics and/or pharmacodynamics. Many therapeutic agents under development are not volatile and have low-molecular-weight (around 1000 amu); these are compounds that are ideally suited for analysis by liquid chromatography, with traditional ultraviolet or fluorescent detectors. However, more recently, combined techniques, such as a liquid chromatography (LC)/mass spectrometry (MS), have considerably improved analytical selectivity and sensitivity. LC/MS/MS methods have facilitated a rapid data turnaround and they also require minimal methods development. Unfortunately, due to the complexity and the protein content of biological fluids, the direct injection of the samples is not compatible with most chromatographic systems. Biological samples are problematic due to the irreversible adsorption of proteins in the stationary phase, resulting in a substantial loss of column efficiency and increase in back pressure.1 In addition, it has become widely recognized that samples of high complexity can result in a matrix effect in mass spectrometry, which leads to an under- or over-estimation of analyte concentrations.2,3Therefore, an appropriate sample preparation is critical and a key consideration for the development of quantitative HPLC methods for measuring drugs in biological fluids. The role of sample preparation also continues to be an important area of development, since the increased acceptance of high-throughput instrumentation such as LC/MS/MS, has shifted the analysis bottleneck backward towards sample preparation.4 In this step the analytical procedures for overcoming the difficulties associated with highly complex and proteineous biological fluids is solved by the simple precipitation of proteins with an organic solvent, with or without the addition of an acid, followed by centrifugation.5,6 The truth is that this procedure provides no real selectivity, and co-precipitation of the analyte, with occlusion in the protein pellet may occur.7 The possibility of signal enhancement/suppression effects in MS due to the precipitation agent also exist. In addition, these ‘quick-and-dirty’ sample preparation techniques, such as protein precipitation or simple ‘dilute-and-shoot’ can result in serious signal matrix effects8 and results in dilution of the analyte. The dilution can be especially problematic when working with low levels of analytes, such as frail bioavailable compounds and metabolites. Sensitivity, selectivity, and sample clean-up can be enhanced by the commonly employed techniques of solid-phase extraction (SPE) or liquid/liquid extraction (LLE).9 These procedures are typically performed off-line, either manually or semi-automatically and may require large volumes of hazardous solvents. Although solvent requirements have been drastically decreased by micro-extraction procedures, such as solid-phase microextraction (SPME)10 or liquid–liquid microextraction,11 automation of these techniques for the liquid chromatographic systems is not common.
Very recently, Rezaee et al. have introduced a novel modality of liquid–liquid microextraction, referred as dispersive liquid–liquid microextraction (DLLME).12 DLLME employs a mixture of a high-density solvent extractant and a water miscible, polar solvent as the disperser. Acetone, methanol and acetonitrile can be used as disperser solvents; whereas chlorinated solvents (e.g. chlorobenzene and chloroform) are useful as extractants. When this solution is added to a sample it forms a cloudy state consisting of fine droplets of extractants dispersed in the matrix. The large contact surface between the sample and the droplets speeds up the mass transference processes. After centrifugation, the extractant solvent settles on the bottom of the vial. Up to now, DLLME has been successfully applied for extraction of organic and inorganic analytes13–26 from water. Recently, some literatures reported the application of DLLME in biological samples.21–23
The goal of this work is to assess whether DLLME can be used as a valuable sample concentration approach for the determination of letrozole in biological fluids and water samples using high-performance liquid chromatography (HPLC) analysis. Letrozole was used as the model compound in the development and evaluation of the procedure. Letrozole, (4,4/-[1H-1,2,4-triazol-1-yl-methylene]bis-benzonitrile), is a selective nonsteroidal inhibitor of the aromatase system (oesterogen synthetase), that is used for the treatment of oesterogen dependency in breast cancer patients.27 The chemical structure of letrozole is shown in Fig. 1. Letrozole is readily and completely absorbed in the gastrointestinal tract. It is slowly metabolized in the liver to an inactive carbinol metabolite, which is then excreted as glucoronide in the urine.28 Several analytical methods have been presented for quantification of letrozole in plasma samples.29–32 The influence of extractant and disperser solvents, as well as ionic strength, on the performance of this method, is discussed in detail later.
 | ||
| Fig. 1 Structure of letrozole. | ||
2. Experimental
2.1. Chemicals and reagents
Letrozole was purchased from Merck Company (Darmstadt, Germany). Carbon tetrachloride (GR), chloroform and chlorobenzene as extraction solvents and acetone, acetonitrile and methanol as dispersive solvents and sodium chloride were obtained from Merck. The water used was purified at Youngling Ultra Pure Water Purification System (Aqua Max™ –Ultra, South Korea).The correct amount of letrozole was dissolved in methanol to obtain the stock solution of the analyte with a concentration of 250 mg L−1. Working standard solutions were freshly prepared by diluting the standard solution of the analyte with ultrapure water to the required concentration. All of the stock solution was stored at 4 °C and kept stable for at least four weeks. Tap water was obtained from our laboratory (Tarbiat Modares University, Tehran, Iran). The urine sample was obtained from a healthy individual and was collected in disposable polyethylene containers and kept at 4 °C before analysis. A frozen human plasma sample was obtained from the Iranian Blood Transfusion Organization (Tehran, Iran), thawed and allowed to reach room temperature.
2.2 HPLC System
Chromatographic separations were carried out on a Varian HPLC equipped with a 9012 HPLC pump (Mulgrave, Australia), a 9010 autosampler (having a 20 μL sample loop) and a Varian 9050 UV/Vis detector. Separations were carried out on a Zorbax Extend C18 column (15 cm × 4.6 mm, with 3 μm particle size) from Agilent Company (Wilmington, DE, USA). A mixture of phosphate buffer (pH = 6.8, 10 mM) and acetonitrile (55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :44) at a flow rate of 1 mL min−1 was used as a mobile phase in isocratic elution mode. The injection volume was 20 μL for all the samples and the detection was performed at a wavelength of 242 nm.
:44) at a flow rate of 1 mL min−1 was used as a mobile phase in isocratic elution mode. The injection volume was 20 μL for all the samples and the detection was performed at a wavelength of 242 nm.
2.3. Dispersive liquid–liquid microextraction procedure
A 10.0 mL portion of double-distilled water was placed in a 40 mL glass tube with conical bottom and spiked at a level of 100 μg L−1 of letrozole. A mixture of the disperser solvent (2.0 mL of acetone) and extraction solvent (142 μL of chloroform) was injected rapidly into the sample solution by using a 5.0 mL gastight syringe. The cloudy solution produced was centrifuged for 5 min at 6000 rpm. After centrifuging, the dispersed fine droplets of chloroform became the sediment at the bottom of the test tube (about 30 ± 2 μL). The sediment phase was completely transferred to another test tube with a conical bottom using a 100 μL HPLC syringe. After the evaporation of the solvent in a water bath the residue was dissolved in 30 μL HPLC grade methanol and injected into the HPLC system. All experiments were performed in triplicate and the mean values of the results were reported.3. Results and discussion
In the present work, DLLME combined with HPLC-UV was developed for extraction and determination of letrozole in biological fluids and water samples. In order to obtain a high recovery as well as preconcentration factor (PF), the effect of different parameters—such as the kind of extraction solvents, disperser solvents plus the volume they accommodated and salt additions—were examined, then optimal conditions were selected. In order to study the explained parameters more easily, extraction recovery (ER) and PF have been calculated based on eqn (1) and eqn (2), and are reported below:| PF = Csed/C0 | (1) |
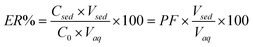 | (2) |
C sed was calculated from a suitable calibration curve that was obtained from the direct injection of letrozole standards in the concentration range of 2–25 mg L−1 into the HPLC.
3.1. Selection of extraction solvent
The proper selection of the extraction solvent is a key step to reaching the optimal conditions of DLLME on biological fluids. Three chlorinated solvents, carbon tetrachloride (CCl4), chloroform (CHCl3) and chorobenzene (C6H5Cl), with densities above 1 g mL−1, possessing low water solubility and different polarities, were considered as extraction solvents. Formation of a sedimentation phase was investigated with 10.0 mL aqueous samples. A series of sample solutions were extracted using 2.0 mL of acetone containing different volumes of extraction solvents to achieve about 30 μL volumes in the sediment phase. Thereby, 71, 76 and 142 μL of chlorobenzene, carbon tetrachloride and chloroform were used, respectively. The average ER% for different extraction solvents were 20.6, 15.6 and 32.6% for chlorobenzene, carbon tetrachloride and chloroform, respectively. The results revealed that chloroform has the highest extraction recovery in comparison with the other tested solvents. It is probably because of the higher solubility of letrozole in chloroform in comparison with chlorobenzene and carbon tetrachloride. Also, the evaporation of chloroform is easier than the other tested solvents. Therefore, chloroform was selected as the extraction solvent.3.2. Selection of disperser solvent
The most important point for the selection of a disperser solvent is its miscibility in organic phase (extraction solvent) and aqueous phase (sample solution). Thereby, acetone, acetonitrile and methanol were selected as disperser solvents. A series of sample solutions containing letrozole were prepared and extracted by using 2.0 mL of the disperser solvent containing 142 μL of chloroform as the extraction solvent. Considering the sedimentation phase volume, it was found that by the combination of chloroform and acetonitrile, the sediment phase volume was remarkably higher than 30 μL and the cloudy state was not formed well, whereas in the case of chloroform and methanol or acetone it was about 30 μL. Therefore, acetone and methanol were selected for further studies. The obtained results indicated that the ER% by using acetone and methanol as the disperser solvents were 34.3 and 33.6% respectively. According to the results, variations of recoveries using different disperser solvents are not remarkable, thus, acetone was selected as the disperser solvent because of its low toxicity as well as its low cost.3.3. Effect of extraction solvent volume
To examine the effect of the extraction solvent volumes, solutions containing different volumes of chloroform were examined with the same DLLME procedures. The experimental conditions are fixed and include the use of 2.0 mL of acetone containing different volumes of chloroform (122, 132, 142, 152 and 162 μL). It is clear that by increasing the volume of chloroform from 122 to 162 μL, the volume of the sediment phase increases from 11 to 57 μL. According to Fig. 2, by increasing the volume of chloroform up to 142 μL, the ER% of the analyte increases. However, due to the increasing of the sediment phase, the volume and dilution are affected and PF decreases (Fig. 2). To obtain a higher PF and ER% value, 142.0 μL of the extraction solvent was used as the optimal volume for the extraction solvent in subsequent experiments.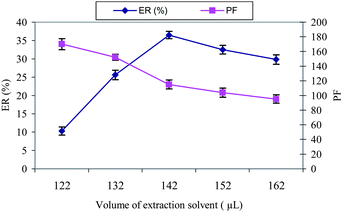 | ||
| Fig. 2 The effect of the extraction solvent (CHCl3) volume on the ER% and PF of letrozole. Extraction conditions: water sample volume, 10.0 mL; disperser solvent (acetone) volume, 2.0 mL; concentration of letrozole, 100 μg L−1. | ||
3.4. Effect of disperser solvent volume
Variation on the volume of acetone as a disperser solvent causes changes in the volume of the sediment phase. To achieve a constant volume of sediment phase, the volume of acetone and chloroform were changed, simultaneously. The experimental conditions were fixed and included the use of different volumes of acetone, 0.5, 1.0, 2.0 and 4.0 mL containing 100, 120, 142, and 158 μL of chloroform, respectively. The results are shown in Fig. 3. According to the curve, the ER% of letrozole increases primarily and then decreases via increasing the volume of acetone. It appears that at low volume, acetone's cloudy state is not well formed, making recovery low. When using higher volumes of acetone, the solubility of letrozole in the water increases, and consequently the ER% decreases. Thus, 2.0 mL of acetone was chosen as the optimum volume in all further studies.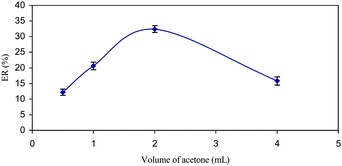 | ||
| Fig. 3 The effect of the disperser solvent (acetone) volume on the ER% of letrozole. Extraction conditions: water sample volume, 10.0 mL; extraction solvent (CHCl3) volumes, 100, 120, 142 and 158 μL; concentration of letrozole, 100 μg L−1. | ||
3.5. Effect of extraction time
In DLLME, extraction time is defined as the interval of time between the injection of the mixture of a disperser and extraction solvents, before starting the centrifuge. According to other literatures,12–26 time has no influence on the ER%, because after the formation of the cloudy solution, the surface area between the extraction solvent and the aqueous phase is very high. Thereby, transition of the analytes from the aqueous phase to extraction solvent is fast. Subsequently, the equilibrium state is achieved quickly so the extraction time is very short. This is the chief advantage of the DLLME technique, time independence. In the present method, the time-consuming step is centrifuging a sample solution in the extraction procedure, this takes about 3 min.3.6. Salt addition
The effect of increasing the ionic strength of the evaluated water sample by adding NaCl at 0–8%, w/v into the water sample spiked with letrozole at a level of 100 μg L−1 was investigated; the DLLME experimental conditions were the same as those described before. It is clear that by increasing the NaCl percentage the volume in the sediment phase increases to 33–55 μL, because of the decrease of solubility of the extraction solvent in the presence of salt. The addition of salt has no effect on the ER%, this is possibly because of two opposite effects of addition of salt in DLLME. One is to increase the volume of settled phase and decrease the dispersion efficiency, which reduces the ER%; another is the salting-out effect, which increases the ER%. Because of the increase in the volume of the sediment phase, the PF decreases (Fig. 4).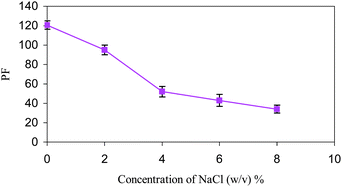 | ||
| Fig. 4 The effect of salt addition on the preconcentration factor of letrozole. Extraction conditions: water sample volume, 10.0 mL; disperser solvent (acetone) volume, 2.0 mL; extraction solvent (CHCl3) volume, 142 μL; concentration of letrozole, 100 μg L−1. | ||
4. Method performance
4.1. Analytical performance
The calibration curve of letrozole with a linear range of 1–500 μg L−1 (nine standards) and a suitable coefficient of determination (r2 = 0.997), were obtained under the optimized condition in aqueous sample. The PF of the proposed method was 120 at a concentration level of 100 μg L−1 of letrozole and a volume of 10.0 mL of aqueous sample. The relative standard deviations (RSD, n = 5) for letrozole extraction and its determination was 3.2% in aqueous sample. The limit of detection (LOD), based on a signal-to-noise ratio (S/N) of 3 was 0.3 μg L−1 in aqueous sample. The calibration graphs were linear in the range of 2.5–500 μg L−1 (eight standards) and 5–500 μg L−1 (seven standards) with detection limits of 0.7 μg L−1 and 1.5 μg L−1 in urine and plasma samples, respectively. The figures of merit of the proposed method are shown in Table 1. LODs between 12.5 and 25 ng mL−1 were reported for the determination of letrozole in urine samples by using a micellar electrokinetic chromatography method.33 Limits of quantitation (LOQs) of letrozole in plasma and urine of 1.40 nmol L−1 and 2.80 nmol L−1, respectively, were reported using automated liquid-solid extraction and fluorescence detection.304.2. Extraction of letrozole from the aqueous samples
In order to study the suitability of the proposed DLLME method for the determination of letrozole in the real samples, the developed technique was applied for the extraction of the drug from a tap water sample. Tap water was spiked with a definite amount of letrozole. Ultimately, the extraction was performed at optimized conditions using the proposed methods; the results are shown in Table 2. Results demonstrated that the tap water matrices, in our present context, had little effect on the DLLME method.4.3. Extraction of letrozole from human urine and plasma samples
Due to the importance of the analysis of letrozole in biological samples,29–32 the proposed method was applied to determine the concentration of letrozole in plasma and urine samples; the obtained results are summarized in Table 2. In order to reduce the matrix effect, the urine sample was diluted to 1:5, using deionized water. It is worthy to note that the spiking of the biological samples was done before the pretreatment of the samples. To the best of our knowledge at the present time, in order to perform the DLLME procedure on plasma samples (to have a relatively clean sediment phase), some extra processes are needed. First, the human plasmas were dissolved in a suitable amount of acetonitrile such as 1:1 (v/v), reducing the matrix effect, and then the mixtures were centrifuged. Second, they were filtered to obtain a clear solution and remove the dirty solution at the bottom of the test tubes. At the end, the clear solutions were diluted to 1:10 for the DLLME procedure.
The chromatograms of the urine samples spiked at a 30 μg L−1 concentration level and the plasma samples spiked at a 50 μg L−1 concentration level of letrozole are shown in Fig. 5a and 5b respectively. The results demonstrate that the urine and plasma matrices had little effect on the DLLME method. The comparison of the proposed method with other reported works29–32 demonstrates that the DLLME-HPLC-UV method has a wide linear range, a lower detection limit and higher preconcentration factor. In addition the method benefits from a very short extraction time, ease of operation in the extraction and determination of letrozole, and is also cheap.
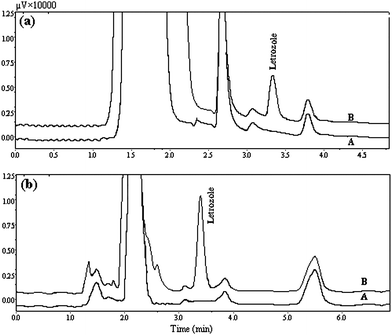 | ||
| Fig. 5 (a) HPLC chromatograms of (A) before spiking the urine with letrozole, (B) 30 μg L−1 urine spiked with letrozole after extraction via proposed method at optimum conditions. (b) HPLC chromatograms of (A) before spiking plasma with letrozole, (B) 50 μg L−1 plasma spiked with letrozole after extraction via proposed method at optimum conditions. Extraction conditions, as in Fig. 4. | ||
5. Conclusions
Dispersive liquid–liquid microextraction combined with HPLC-UV detection tackles the determination of letrozole in biological fluids and water samples in a simple way. The method is simple, rapid and inexpensive. A high preconcentration factor was obtained easily through this method and a detection limit at a 0.3 μg L−1 level was achieved with only 10.0 mL of aqueous sample. In this method sample preparation time as well as the consumption of toxic organic solvents were minimized without affecting the sensitivity of the method. Although the obtained results in this work are related to determination of letrozole, the system could be readily applied for the determination of other drugs, using different analytical instruments.Acknowledgements
Financial support from Tarbiat Modare University is gratefully acknowledged.References
- S. Souverain, S. Rudaz and J.-L. Veuthey, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2004, 801, 141–156 CrossRef CAS.
- B. K. Matuszewski, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2006, 830, 293–300 CrossRef CAS.
- B. K. Matuszewski, M. L. Constanzer and C. M. Chavez-Eng, Anal. Chem., 2003, 75, 3019–3030 CrossRef CAS.
- M. Berna, A. T. Murphy, B. Wilken and B. Ackermann, Anal. Chem., 2002, 74, 1197–1201 CAS.
- A. L. Jayewardene, B. Kearney, J. A. Stone, J. G. Gambertoglio and F. T. Aweeka, J. Pharm. Biomed. Anal., 2001, 25, 309–317 CrossRef CAS.
- R. Gage and D. A. Stopher, J. Pharm. Biomed. Anal., 1998, 17, 1449–1453 CrossRef CAS.
- C. Schafer and D. Lubda, J. Chromatogr., A, 2001, 909, 73–78 CrossRef.
- C. Polson, P. Sarkar, B. Incledon, V. Raguvaran and R. Grant, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2003, 785, 263–275 CrossRef CAS.
- F. F. Cantwell and M. Losier, Sampling and Sample Preparation for Field and Laboratory, ed. J. Pawliszyn, Elsevier, New York, 2002, pp. 297–340 Search PubMed.
- G. Theodoridis and G. J. de Jong, Adv Chromatogr., 2005, 43, 231–271 CAS.
- M. Ma and F. F. Cantwell, Anal. Chem., 1998, 70, 3912–3919 CrossRef CAS.
- M. Rezaee, Y. Assadi, M. R. Milani Hosseini, E. Aghaee, F. Ahmadi and S. Berijani, J. Chromatogr., A, 2006, 1116, 1–9 CrossRef CAS.
- S. Berijani, Y. Assadi, M. Anbia, M. R. Milani Hosseini and E. Aghaee, J. Chromatogr., A, 2006, 1123, 1–9 CrossRef CAS.
- R. Rahnama Kozani, Y. Assadi, F. Shemirani, M. R. Milani Hosseini and M. R. Jamali, Talanta, 2007, 72, 387–393 CrossRef CAS.
- R. Rahnama Kozani, Y. Assadi, F. Shemirani, M. R. Milani Hosseini and M. R. Jamali, Chromatographia, 2007, 66, 81–86 CrossRef CAS.
- N. Fattahi, Y. Assadi and M. R. Milani Hosseini, J. Chromatogr., A, 2007, 1157, 23–29 CrossRef CAS.
- A. Bidari, E. Zeini Jahromi, Y. Assadi, M. R. Milani Hosseini and M. R. Jamali, Microchem. J., 2007, 87, 6–12 CrossRef CAS.
- E. Zeini Jahromi, A. Bidari, Y. Assadi, M. R. Milani Hosseini and M. R. Jamali, Anal. Chim. Acta, 2007, 585, 305–311 CrossRef.
- H. Jiang, Y. Qin and B. Hu, Talanta, 2008, 74, 1160–1165 CrossRef CAS.
- M. T. Naseri, P. Hemmatkhah, M. R. Milani Hosseini and Y. Assadi, Anal. Chim. Acta, 2008, 610, 135–141 CrossRef.
- H. Xu, D. Song, Y. Cui, S. Hu, Q. W. Yu and Y. Q. Feng, Chromatographia, 2009, 70, 775–781 CrossRef CAS.
- L. Lili, H. Xu, D. Song, Y. Cui, S. Hu and G. Zhang, J. Chromatogr., A, 2010, 1217, 2365–2370 CrossRef.
- M. B. Melwanki, W. S. Chen, H. Y. Bai, T. Y. Lin and M. R. Fuh, Talanta, 2009, 78, 618–622 CrossRef CAS.
- D. Nagaraju and S. D. Huang, J. Chromatogr., A, 2007, 1161, 89–97 CrossRef CAS.
- L. Farina, E. Boido, F. Carrau and E. Dellacassa, J. Chromatogr., A, 2007, 1157, 46–50 CrossRef CAS.
- M. A. Farajzadeh, M. Bahram and J. A. Jonsson, Anal. Chim. Acta, 2007, 591, 69–79 CrossRef CAS.
- H. M. Lamb and J. C. Adkins, Drugs, 1998, 56, 1125–1140 CrossRef CAS.
- T. J. Iveson, I. E. Smith, J. Ahern, D. A. Smithers, P. F. Trunet and M. Dowsett, J. Clin. Endocrinol. Metab., 1993, 77, 324–331 CrossRef CAS.
- C. U. Pfister, M. Duval, J. Godbillon, G. Gosset, D. Gygax, F. Marfil, A. Sioufi and B. Winkler, J. Pharm. Sci., 1994, 83, 520–524 CrossRef CAS.
- F. Marfil, V. Pineau, A. Sioufi and S. J. Godbillon, J. Chromatogr., B: Biomed. Sci. Appl., 1996, 683, 251–258 CrossRef CAS.
- A. Sioufi, N. Gauducheau, V. Pineau, A. Marfil, A. Jaouen, J. M. Cardot, S. J. Godbillon, C. Czendilk, H. Howald, C. U. Pfister and F. Vreeland, Biopharm. Drug Dispos., 1997, 18, 779–789 CrossRef CAS.
- E. R. Trosken, K. Fischer, W. Volkel and W. K. Lutz, Toxicology, 2006, 219, 33–40 CrossRef.
- J. Rodriguez Flores, A. M. Salcedo, M. J. Llerena and L. M. Fernandez, J. Chromatogr., A, 2008, 1185, 281–290 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |