Quantitative NMR spectroscopy for the rapid measurement of methylglyoxal in manuka honey
James A.
Donarski
,
Dominic P. T.
Roberts
and
Adrian J.
Charlton
*
The Food and Environment Research Agency, Sand Hutton, York, YO41 1LZ, UK. E-mail: adrian.charlton@fera.gsi.gov.uk
First published on 11th August 2010
Abstract
The accurate and rapid measurement of methylglyoxal in honey at concentrations applicable to those found in manuka honey using qNMR is reported. The qNMR method gave comparable results to those obtained by derivatisation of methylglyoxal with ortho-phenylenediamine and subsequent LC-MS or LC-UV detection. Uniquely, the qNMR method is performed directly on the diluted honey without chromatographic separation, sample derivatisation or generation of a calibration series.
1. Introduction
Manuka honey is derived from bees that forage on the manuka tree (Leptospermum scoparium) and has been demonstrated to exhibit significant non-peroxide antibacterial activity.1 It is classified according to its antimicrobial “strength” using the Unique Manuka Factor (UMF). This is determined by comparing the size of the zone of inhibition of Staphylococcus aureus induced by the honey with that induced by phenol solutions. Manuka honey that demonstrates an antibacterial activity equivalent to a 5% solution of phenol is classified as UMF 5+ and this principle applies up to the comparison with a 30% phenol solution at which concentration a manuka honey would be rated as UMF 30+.2 It has recently been demonstrated that this non-peroxide antimicrobial activity is due to the presence of methylglyoxal (2-oxopropanal, Fig. 1). | ||
| Fig. 1 Chemical structure of methylglyoxal (1), methylglyoxal monohydrate (2) and methylglyoxal dihydrate (3). | ||
Methylglyoxal is a naturally occurring dicarbonyl compound. It is formed by several biological pathways,3 during the caramelisation of carbohydrates and during the Maillard reaction between carbohydrates and amino acids.4 Methylglyoxal undergoes a “spontaneous” reaction with water and it has been estimated that in aqueous solution less than 1% of the methylglyoxal remains unreacted.5 It predominantly forms two compounds: methylglyoxal monohydrate and methylglyoxal dihydrate6 (Fig. 1). The reactivity of methylglyoxal, its low molecular mass and lack of ultraviolet (UV) chromophore have made direct detection of methylglyoxal in manuka honey problematic using conventional analytical detection systems. Therefore, methods to detect methylglyoxal have usually employed derivatisation with ortho-phenylenediamine (OPD) to form the 2-methylquinoxaline.7–9 The derivatisation step must be performed in a controlled manner as OPD can catalyse the formation of methylglyoxal by the Maillard reaction10 leading to overestimates of concentration. A method for direct quantification of methylglyoxal in manuka honey has been demonstrated by Adams et al.11 using an HPLC system with mixed mode size exclusion/ligand exchange columns connected in series. Quantification of methylglyoxal was performed using a refractive index (RI) detector. Using this method, methylglyoxal partially coeluted with fructose and in the case of several non-manuka honeys, falsely high methylglyoxal levels were determined because an unidentified compound eluted at the same retention time as methylglyoxal.
Here we describe the direct determination of methylglyoxal in manuka honey using the primary ratio method, quantitative NMR spectroscopy (qNMR). The validity of using qNMR was confirmed by generating and analysing a series of methylglyoxal spiked honey and aqueous samples. qNMR was then used to determine the concentration of methylglyoxal in 6 retail manuka honey samples and the results were compared with those obtained using OPD derivatisation, liquid chromatography (LC) separation and then either mass spectrometric or UV detection.
2. Experimental
2.1. Materials
Six commercial manuka honeys, with UMF factors (as declared on the label) ranging between 5 and 25 and two non-active honeys were purchased from local retail outlets. The sample information is listed in Table 1.All chemicals used were of a purity of ≥97% unless otherwise stated. Acetic acid and formic acid (approximately 85% pure) were supplied by Fischer Scientific (UK). Deuterium oxide (2H2O) was supplied by Goss Scientific Instruments Ltd (UK). 3-trimethylsilyl[2,2,3,3-D4]propionic acid (TSP) was supplied by Avocado Research Chemicals Ltd (UK). Methylglyoxal (approximately 40% wt in water), 2-methylquinoxaline, o-phenylenediamine, sodium azide and sodium di-hydrogen phosphate were supplied by Sigma-Aldrich Co. (UK). Di-potassium hydrogen phosphate and di-hydrogen potassium phosphate were supplied by BDH Chemicals Ltd (UK). Purine and hexakis(1H,1H,3H-tetrafluoro-pentoxy)-phosphazene were supplied by Agilent Technologies (USA). Ultrapure water was obtained from an Elga Option 2 water purifier.
2.2. NMR methods
Honey was diluted with ultrapure water to 75 mg mL−1 ± 3.75 mg mL−1. The diluted standards were centrifuged to remove any remaining suspended material prior to filtration through a 0.2 µm PTFE syringe filter. The standards were prepared for NMR spectroscopy by adding 200 µL of the filtered honey solution to 60 µL of sodium azide solution (10 mM dissolved in 2H2O), 60 µL of phosphate buffer solution (250 mM, pH 6.9 containing 380 µM TSP dissolved in 2H2O), to this solution was added either 0, 50, 100, 150, or 250 µL of 95 ± 5 mg L−1 methylglyoxal and finally the volume was made to 600 µL using ultrapure water to generate the matrix matched calibration series. The standards were thoroughly mixed using a vortex mixer prior to 1H NMR spectroscopic analysis.
Furthermore, a calibration series for methylglyoxal in ultrapure water was prepared as detailed above by substituting filtered honey solution with ultrapure water.
The precision (repeatability) of the method was assessed by preparing a sample of honey G, spiked at two concentrations of methylglyoxal, 11 and 34 mg L−1. Eight replicates were analysed and quantified by qNMR (Section 2.2.2).
The accuracy was assessed by preparing samples of honey G, spiked at 11 and 34 mg L−1, in triplicate. The samples were quantified by qNMR and the Z scores calculated using eqn (1).
 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 536) were acquired with a spectral width of 13.9991 ppm, giving an acquisition time of 4.6794 s. A recycle time of 14.7 s was determined experimentally to produce quantitative data with optimized sensitivity. A sine bell-shaped window function phase shifted by 90° was applied over all data points before Fourier transformation, phase, and baseline correction. The chemical shift of all data was referenced to the TSP resonance at 0 ppm. All spectra were acquired at 300 K.
536) were acquired with a spectral width of 13.9991 ppm, giving an acquisition time of 4.6794 s. A recycle time of 14.7 s was determined experimentally to produce quantitative data with optimized sensitivity. A sine bell-shaped window function phase shifted by 90° was applied over all data points before Fourier transformation, phase, and baseline correction. The chemical shift of all data was referenced to the TSP resonance at 0 ppm. All spectra were acquired at 300 K.
Eight unrecorded and 256 acquisition transients were collected giving a total experimental time of approximately 1 hour 5 minutes.
To determine the concentration of methylglyoxal in each sample, the methyl peaks of methylglyoxal monohydrate (2.297–2.314 ppm, 3H, s), methylglyoxal dihydrate (1.369–1.386 ppm, 3H, s) and TSP (−0.01 to 0.01 ppm, 9H, s) were integrated. The concentration of methylglyoxal in the NMR sample was calculated using eqn (2).
 | (2) |
 | (3) |
2.3. MGO determination using LC-MS
Liquid chromatography time-of-flight mass spectrometry (LC-TOF-MS) was performed using a method based on that of Lo et al.7 with a slight modification to the derivatisation procedure.Sample derivatisation was performed by adding 800 µL of honey solution to 200 µL OPD (60 mg mL−1 dissolved in ultrapure water). The samples were thoroughly mixed using a vortex mixer prior to heating at 60 °C for 30 minutes. Samples were cooled to room temperature and dissolved to 10 mL using ultrapure water before analysis. Standards were derivatised as detailed above by substituting honey solution with methylglyoxal standard.
2.4 MGO determination using LC-UV
LC-UV analysis was performed using a method based on that of Mavric et al.8A range of 2-methylquinoxaline calibration standards ranging from 1 to 100 µg mL−1 were prepared in 0.5 M sodium phosphate solution (pH 6.5).
3. Results and discussion
3.1 NMR calibration series
The 1H NMR spectrum of methylglyoxal dissolved to a concentration of 22.5 mg L−1 in ultrapure water is shown in Fig. 2. The spectrum predominantly contained two singlet resonances at 1.378 and 2.306 ppm. These corresponded to the methyl hydrogen atoms present in methylglyoxal di- and monohydrate, respectively. The resonance at 5.287 was assigned to the alkyl proton of methylglyoxal monohydrate. The alkyl proton of methylglyoxal dihydrate resonates at 4.806 ppm and is not observed because of the presaturation conditions used. The resonances at 1.923 and 8.458 ppm were assigned to acetic and formic acid, respectively. Resonance assignments were confirmed by over spiking experiments. | ||
| Fig. 2 500 MHz 1H NMR spectrum of 22.5 mg L−1 methylglyoxal. | ||
The calibration graph demonstrating the linearity of the NMR response with increasing methylglyoxal concentration in matrix matched honey and water matrix samples is shown in Fig. 3. The calculated values were determined using eqn (2) and the expected values were determined from the masses used assuming a purity of the methylglyoxal standard of 40%. Non-active honey G was observed to contain a low concentration of naturally occurring methylglyoxal that was calculated to be approximately 28 mg kg−1. Non-active honey H was observed to contain an unidentified compound that resonated at 1.374 ppm that was included in the integration range chosen. For the calibration series, the influence of the naturally occurring methylglyoxal in honey G and the interfering peak in honey H were subtracted from their respective matrix matched calibration series using the integral values determined from the non-spiked honeys. A linear regression line constrained to the origin was calculated, the gradient of this line was 0.9786 and its R2 value was 0.996. These results confirm that the concentration of methylglyoxal can be accurately determined using qNMR.
 | ||
| Fig. 3 Concentration of methylglyoxal determined by qNMR compared to actual concentration of methylglyoxal determined using sample mass in ultrapure water (◇), honey G (x), and honey H (■). | ||
The expected concentration of methylglyoxal in the NMR sample was calculated assuming a methylglyoxal purity of 40%, although from the specification supplied with the methylglyoxal this purity is only an approximation. The results shown in Fig. 3 were used to determine the accurate purity of the methylglyoxal standard. The purity of the methylglyoxal standard was calculated for each sample used in the calibration series by qNMR. The mean value for the purity was calculated to be 39.05% ± 3.26% (95.4% confidence interval).
The precision of the method was evaluated by performing 8 replicate analyses of a sample of honey G spiked at 11 and 34 mg L−1 (corresponding to methylglyoxal levels in undiluted honey of 440 and 1360 mg kg−1). The relative standard deviation expressed as a percentage was 5.0% and 1.9% for the samples spiked at 11 and 34 mg L−1, respectively. The accuracy was evaluated by preparing, in triplicate, samples spiked at 11 and 34 mg L−1. Z scores were in the range 0.14–0.53 and 0.1–0.53 for the samples spiked at 11 and 34 mg L−1, respectively. The method was proven to be both accurate and precise.
3.2 Methylglyoxal determination in UMF rated honeys
The 1H NMR spectrum of manuka honey obtained from sample 1 is shown in Fig. 4. The full spectrum is shown in the upper window and an expanded region showing the presence of the methylglyoxal mono- and dihydrate and the integral regions chosen is shown in the lower window.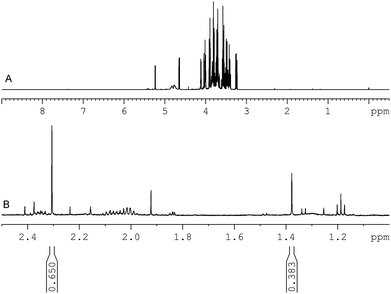 | ||
| Fig. 4 500 MHz 1H NMR spectrum acquired from manuka honey sample A. (a) Spectral region 0.5–9.0 ppm and (b) spectral region 1.0–2.5 ppm showing the methyl resonances of methylglyoxal mono (2.309) ppm and di-hydrate (1.377 ppm) used for quantification. The relative area of these resonances compared to the internal standard is given below each resonance. | ||
The methylglyoxal concentration of manuka honey determined using qNMR, LC-TOF-MS and LC-UV is presented in Table 2.
From the results obtained it can be observed that they are all approximately in agreement although those from qNMR are in most cases lower than those reported by OPD derivatisation. The origin of methylglyoxal in manuka honey has recently been attributed to the presence of dihydroxyacetone in the nectar of manuka flowers.12 The spectra confirmed the presence of dihydroxyacetone in all manuka honey samples studied by NMR although it was not possible to determine the exact concentration without further method development because dihydroxyacetone resonated at 4.423 ppm. This resonance was attenuated by the presaturation conditions used to suppress the residual water peak during NMR data acquisition. A possible explanation of the higher results obtained from derivatisation of methylglyoxal in manuka honey could be the formation of methylglyoxal via the Maillard reaction between sugars and OPD or the creation of methylglyoxal from dihydroxyacetone due to the elevated temperature required for derivatisation.
4. Conclusion
We have successfully developed a qNMR method for the quantification of methylglyoxal in manuka honey. In solution, methylglyoxal is present as the mono- and dihydrate. The qNMR method presented here has independently measured the concentration of methylglyoxal mono- and dihydrate, summing these values to determine the methylglyoxal concentration. Thus, for the first time the concentration of methylglyoxal has been determined in manuka honey, without the need for chromatographic separation or a derivatisation procedure. This approach has shown that previously applied methodology may overestimate the MGO concentration in manuka honey.The results demonstrate significant concentrations of methylglyoxal in manuka honey. Methylglyoxal concentrations have been previously determined in a variety of foodstuffs: brewed coffee (7 mg L−1),13 cheese (11 mg kg−1),14 high energy carbonated soft drinks (1 mg L−1),7 wine (25 mg L−1)9 and cookies (81 mg kg−1).15 Although these are significantly lower than the concentrations determined in manuka honey.
Gut microflora is affected by methylglyoxal16 and its presence may significantly impact on absorption in the body by processes such as sequestration and chemical reaction. Campbell et al. showed that methylglyoxal was bacteriostatic, being able to arrest the growth of Escherichia coli without killing the bacteria. This result may tie in with the anecdotal, although recently refuted,17 evidence relating to the beneficial effects of manuka honey towards suppression of gastrointestinal conditions, which may be related to the antimicrobial activity of the honey imparted by high concentrations of methylglyoxal.
Acknowledgements
The authors thank the Interact Partnership (a partnership of six UK government research organisations, including Fera) for funding this work. The Interact Partnership is funded by the department for Business, Innovation and Skills.References
- M. J. Snow and M. Manley-Harris, Food Chem., 2004, 84, 145–147 CrossRef CAS.
- K. L. Allen, P. C. Molan and G. M. Reid, J. Pharm. Pharmacol., 1991, 43, 817–822 CAS.
- M. P. Kalapos, Toxicol. Lett., 1999, 110, 145–175 CrossRef CAS.
- P. J. Thornalley, A. Langborg and H. S. Minhas, Biochem. J., 1999, 344(Pt 1), 109–116 CrossRef CAS.
- T. W. Lo, M. E. Westwood, A. C. McLellan, T. Selwood and P. J. Thornalley, J. Biol. Chem., 1994, 269, 32299–32305 CAS.
- I. Nemet, D. Vikic-Topic and L. Varga-Defterdarovic, Bioorg. Chem., 2004, 32, 560–570 CrossRef CAS.
- C. Y. Lo, S. M. Li, Y. Wang, D. Tan, M. H. Pan, S. M. Sang and C. T. Ho, Food Chem., 2008, 107, 1099–1105 CrossRef CAS.
- E. Mavric, S. Wittmann, G. Barth and T. Henle, Mol. Nutr. Food Res., 2008, 52, 483–489 CrossRef CAS.
- A. C. da Silva Ferreira, S. Reis, C. Rodrigues, C. Oliveira and P. G. de Pinho, J. Food Sci., 2007, 72, S314–S318 CrossRef.
- Y. Wang and C. T. Ho, Food Chem., 2008, 109, 1–3 CrossRef CAS.
- C. Adams, C. H. Boult, B. J. Deadman, J. M. Farr, M. N. C. Grainger, M. Manley-Harris and M. J. Snow, Carbohydr. Res., 2008, 343, 651–659 CrossRef CAS.
- C. J. Adams, M. Manley-Harris and P. C. Molan, Carbohydr. Res., 2009, 344, 1050–1053 CrossRef CAS.
- M. Nagao, Y. Fujita, K. Wakabayashi, H. Nukaya, T. Kosuge and T. Sugimura, Environ. Health Perspect., 1986, 67, 88–91.
- W. Bednarski, L. Jedrychowski, E. G. Hammond and Z. L. Nikolov, J. Diary Sci., 1989, 72, 2474–2477 CrossRef CAS.
- G. Arribas-Lorenzo and F. J. Morales, J. Agric. Food Chem., 2010, 58, 2966–2972 CrossRef CAS.
- A. K. Campbell, R. Naseem, I. B. Holland, S. B. Matthews and K. T. Wann, Arch. Biochem. Biophys., 2007, 468, 107–113 CrossRef CAS.
- A. Wallace, S. Eady, M. Miles, H. Martin, A. McLachlan, M. Rodier, J. Willis, R. Scott and J. Sutherland, Br. J. Nutr., 2010, 103, 1023–1028 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |