2,2′-(1,3-Propanediylbisnitriloethylidine)bis-hydroquinone/TiO2 nanoparticles modified carbon paste electrode for selective determination of dopamine in the presence of uric acid and tryptophan
Mohammad
Mazloum-Ardakani
*a,
Rezvan
Arazi
a,
Hadi
Beitollahi
a and
Hossein
Naeimi
b
aDepartment of Chemistry, Faculty of Science, Yazd University, Yazd, 89195-741, I. R. Iran
bDepartment of Chemistry, Faculty of Science, University of Kashan, Kashan, 87317, I. R. Iran
First published on 28th May 2010
Abstract
A modified carbon paste electrode with 2,2′-(1,3-propanediylbisnitriloethylidine)bis-hydroquinone (PBNBH) and TiO2 nanoparticles has been fabricated and used to study the catalytic oxidation of dopamine (DA), uric acid (UA) and tryptophan (Trp). The overpotential of DA reduced about 0.36 V. Based on its strong catalytic function toward the oxidation of DA, UA and Trp, the modified electrode resolved the overlapping voltammetric response of DA, UA and Trp into three well-defined voltammetric peaks with square wave voltammetry (SWV), which can be used for the simultaneous determination of these species in a mixture. The catalytic peak currents obtained from SWV were linearly dependent on the DA concentration in the range 2.0–10.0 μM and 10.0–1000.0 μM with correlation coefficients of 0.9993 and 0.9998, respectively. The detection limit (2σ) for DA was 0.47 μM. The modified electrode showed good sensitivity and selectivity and has been applied to the determination of DA, UA and Trp simultaneously in real samples with satisfactory results.
1. Introduction
The emergence of nanotechnology has opened new opportunities for the application of nanoparticles in analytical chemistry.1 The similar dimensions of nanoparticles and protein molecules suggest that the integration of the two will lead to attractive new materials. The fascinating physical and chemical properties of nanoparticles (such as colloidal gold and semiconductor quantum-dot nanoparticles), compared to their bulk counterparts, offer excellent prospects for a wide range of biosensing applications.2Dopamine (DA), the most significant among the class of catecholamines, plays an important role in the function of the central nervous, cardiovascular, renal and hormonal systems.3 The determination of DA is a subject of great importance for investigating its physiological functions and diagnosing nervous diseases resulting from DA abnormal metabolism, such as epilepsy, senile dementia, Parkinsonism, schizophrenia and HIV infection.4 However, a major problem in its determination is the interference from ascorbic acid (AA),5 which largely coexists with DA in brain issue. At traditional solid electrodes, AA is oxidized at potentials close to that of DA, which result in overlapped voltammetric responses making their discrimination highly difficult.5 Moreover, the solid electrodes very often suffer from the fouling effort due to the accumulation of oxidized products on the electrode surface, which results in rather poor selectivity and sensitivity. Therefore, the development of new electrochemical sensors for DA has received much interest in the last few decades.6
Uric acid (UA) is the principal final product of purine metabolism in the human body.7 It has been shown that extreme abnormalities of UA levels are symptoms of several diseases (e.g. gout, hyperuricaemia and Lesch–Nyhan syndrome).8,9 Other diseases, such as leukemia and pneumonia are also associated with enhanced urate levels.9 In general, electroactive UA can be irreversibly oxidized in aqueous solution and the major product is allantoin.10 Since UA, AA and DA usually coexist in physiological samples, it is essential to develop simple and rapid methods for their determination in routine analysis. Simultaneous detection of UA and neurotransmitters, especially DA is a problem of critical importance not only in the field of biomedical chemistry and neurochemistry but also for diagnostic and pathological research. Among many methods for determination of DA and UA in biological samples, electrochemical techniques with modified electrodes have been shown to be powerful tools due to their advantages of simple, inexpensive and fast analysis in combination with high sensitivity and selectivity.11 Thus, different kinds of modified electrodes have been fabricated for detection of DA and UA.12–13 All the modified electrodes have some advantages and limitations. Up to now, still there is an expanding demand for the development of simple, reliable and efficient sensors with enhanced characteristics for effective sensing of DA and UA simultaneously.
Tryptophan (Trp) is an essential amino acid with diverse physiological roles, functioning both independently or via incorporation into the structure of larger molecules or polymers, such as proteins. It is a precursor for biologically important molecules, such as the neurotransmitter serotonin and the neurohormone melatonin.14 Abnormal levels of serotonin and melatonin have been shown to be associated with depression15 and Alzheimer's and Parkinson's diseases,16 respectively. It has been shown that the control of dietary intake of Trp (through food or supplements) and the resulting physiological concentrations in the human body has had a positive effect on the regulation of the synthesis of serotonin.17
The regulation of the synthesis of serotonin leads to the controlled synthesis of melatonin18 which promotes sleep. Following the lifting of the USA food and drug association ban on l-Trp, the amino acid is increasingly available in food supplement form, and in some formulations co-administered with melatonin. Given the far reaching role of this amino acid, methods for the detection of Trp in food processing, pharmaceutical formulations and in biological fluids are of importance.19
Electrochemical methods of detecting Trp20,21 have shown promise compared to standard chromatographic22 and electrophoretic23 methods. However, electrochemical detection of Trp at unmodified electrode surfaces is not optimal owing to sluggish electron transfer processes,19 and in complex matrices is subject to interference from molecules with overlapping potentials.
The concept of surface modification of electrodes is one of the exciting developments in the field of electroanalytical chemistry. The modification may lead to significant reduction of overpotentials and increases in the electron transfer rate constant for desired redox reactions at the electrode.24–28 Among them, carbon- based electrodes are especially interesting since their surface can be covalently grafted by various molecules, resulting in a versatile interface.
To the best of our knowledge, no study has been published so far on the simultaneous determination of DA, UA and Trp by using TiO2 nanoparticles modified carbon paste electrode. Thus, here we report the preparation and application of a PBNBH (Scheme 1)/TiO2 nanoparticle modified carbon paste electrode (PBNBH-TNMCPE) as a new electrode during the electrocatalytic procedure and the determination of DA in an aqueous buffer solution. The analytical performance of the modified electrode has also been evaluated during DA-quantification in the presence of UA and Trp.
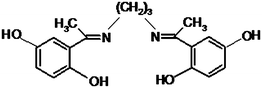 | ||
| Scheme 1 Structure of 2,2′-(1,3-propanediylbisnitriloethylidine)bis-hydroquinone. | ||
2. Experimental
2.1. Chemicals
The DA, UA and Trp were purchased from Merck (Darmstadt, Germany). All other reagents used in this study were all of analytical grade. Graphite powder (Merck) and paraffin oil (DC 350, Merck, density = 0.88 g cm−3) were used as binding agents for the graphite pastes. The buffer solutions were prepared from ortho-phosphoric acid and its salts in the pH range from 2.0–11.0. The experimental results were obtained at room temperature.2.2. Apparatus
SAMA 500 electrochemical work station (Sama instruments, Iran) equipped with a personal computer was used for electrochemical measurements and treating data. The Sama software was used for control and data acquisition. A three electrode electrochemical cell was employed. PBNBH-TNMCPE, saturated calomel electrode (SCE) and platinum wire were used as working, reference and auxiliary electrodes. All potentials reported were versus the SCE. A Metrohm 691 pH/ion meter was also used for pH measurements.2.3. Typical procedure for preparation of 2,2′-(1,3-propanediylbisnitriloethylidine)bis-hydroquinone
To a mixture of 2,5-dihydroxyacetophenone (0.3 g, 2 mmol) in methanol was added 1,3-diaminopropane (0.074 g, 1 mmol) by stirring in one portion. The mixture stirring is continued for 40 min. The progress of the reaction was monitored by TLC. After the completion of the reaction, a yellow substance was precipitated. The solid product was filtered off and washed with cold methanol. The obtained crude product was recrystallized in methanol and the 2,2′-(1,3-propanediylbisnitriloethylidine)bis-hydroquinone as yellow crystalline with m.p. 230–232 °C in 97% yield was obtained.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1562, 1419, 1312, (s, Ar CC), 1219 (s, C–O), 965, 840, 799(s).
N), 1562, 1419, 1312, (s, Ar CC), 1219 (s, C–O), 965, 840, 799(s).
1H NMR (400 MHz/DMSO)/δ ppm: 1.85 (m, 2 H, CH2), 2.1 (s, 6 H, 2 CH3), 3.44 (t, 4 H, 2 CH2), 6.44 (d, 2 H, Ar), 6.56 (d of d, 2 H, Ar), 6.8 (d, 2 H, Ar), 8.4 (s, 2 H, OH), 15.2 (s, br, 2 H, OH).
13C NMR (100 MHz/DMSO)/δ ppm: 16.1, 32.7, 48.4, 115.2, 119.53, 120.7, 121.6, 149.7, 157.0, 173.0.
2.4. Preparation of TiO2 nanoparticles
Colloidal suspension of TiO2 nanoparticles was synthesized by mixing titanium tetraisopropoxide, H2O2, and H2O with volume proportions of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90:200, respectively. The resulting solution was refluxed for 10 h to promote the crystallinity (surface area = 84 m2 g−1 and particle size = 6.7 nm).
:90:200, respectively. The resulting solution was refluxed for 10 h to promote the crystallinity (surface area = 84 m2 g−1 and particle size = 6.7 nm).
2.5. Preparation of electrode
0.005 g of PBNBH was dissolved in CH3Cl and hand mixed with 0.02 g TiO2 nanoparticles and 0.0475 g graphite powder with a mortar and pestle. The solvent was evaporated by stirring. Paraffin were added to the above mixture using a 4 mL syringe and mixed for 20 min until a uniformly-wetted paste was obtained. The paste was then packed into the end of a glass tube (ca. 3.4 mm i.d. and 10 cm long). Electrical contact was made by pushing a copper wire down the glass tube into the back of the mixture. When necessary, a new surface was obtained by pushing an excess of paste out of the tube and polishing it on a weighing paper.3. Results and discussion
3.1. Electrochemical behavior of PBNBH-TNMCPE
The PBNBH complex is insoluble in aqueous solutions, therefore, it can be used in the carbon paste without leaching out from the electrode surface, which leads to a stable chemically modified electrode. The cyclic voltammogram of PBNBH-TNMCPE exhibits an anodic peak at the forward scan, which is related to the oxidation of PBNBH to the quinone form of PBNBQ (Fig. 1). Reduction of quinone form PBNBQ to PBNBH occurs during the reverse scan (cathodic current peak). The peak separation potential, ΔEp was greater than the expected one (59/n) mV for a reversible system. This suggests a quasi reversible behavior in an aqueous medium.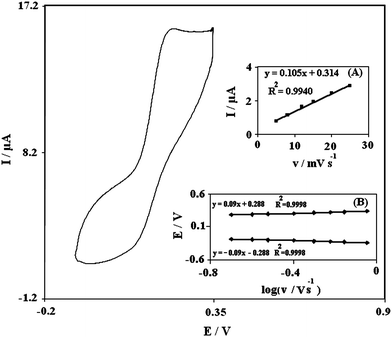 | ||
| Fig. 1 Cyclic voltammograms of PBNBH-TNMCPE in 0.1 M phosphate buffer solution (pH 7.0) at scan rate of 100 mV s−1. Insets: (A) Variations of I versus scan rates. (B) Variation of E versus the logarithm of the scan rates for high scan rates. | ||
In addition, the effect of the potential scan rate on electrochemical properties of the PBNBH/PBNBQ redox couple in PBNBH-TNMCPE was studied in an aqueous solution with cyclic voltammetry. Plots of the peak currents (Ip) were linearly dependent on the scan rates from 50 to 400 mV s−1. A linear correlation was obtained between peak currents, and the scan rate indicates that the nature of the redox process was controlled in a diffusionless manner (Fig. 1A).
Laviron29 derived general expressions for the linear potential sweep voltammetric response for the case of surface confined electroactive species with a small enough concentration:
| Epa = E0 + A ln [1 − α/m] | (1) |
| Epc = E0 + B ln [α/m] | (2) |
For ΔEp > 200/n mV
| log ks = α log (1 − α) + (1 − α) log α − log (RT/nFυ) − α(1 − α)nFΔEp/2.3RT | (3) |
From these expressions, it is possible to determine the transfer coefficient (α) by measuring the variation of the peak potentials with scan rate (υ) as well as the apparent charge transfer rate constant (ks) for electron transfer between the electrode and the surface confined material.
A plot of Ep as a function of logυ yields one straight line with slope equal to 2.3RT/(1 − α)nF for the anodic peak. Inset B of Fig. 1. shows the magnitudes of anodic peak potential as a function of the potential scan rate. We have found that for scan rates above 400 mV s−1, the values of Epa was proportional to the logarithm of scan rate as was indicated by Laviron. The plot is shown in Fig. 1B. Using such a plot and eqn (3), the values of α and ks were 0.34 and 1.55 s−1, respectively.
The values for the surface concentration (Γ) of the modified carbon paste electrode (MCPE), given in mol cm−2 were obtained from following equation.30
| Ip = n2F2AΓυ/4RT | (4) |
3.2. Effect of pH on the electrochemical behaviors of the PBNBH-TNMCPE
The effect of the medium's pH on the electrochemical signal of PBNBH-TNMCPE was investigated. The pH of solutions was adjusted using buffer solutions in the range of 2.0–11.0. Results showed that the peak potential changed over the pH range 2.0–11.0. There was a linear range, which is described by the following equation:| Ep = −0.053 pH + 0.5483 R2 = 0.9994 | (5) |
Such behavior suggests that it obeys the Nernst equation for a two electron and two proton transfer reaction.31
3.3. Electrocatalytic oxidation of DA at PBNBH-TNMCPE
Fig. 2 shows the cyclic voltammetric responses from the electrochemical oxidation of 0.6 mM DA at bare-CPE (Fig. 2A, curve a), TiO2 nanoparticles carbon paste electrode (TNCPE) (Fig. 2A, curve b), PBNBH modified carbon paste electrode (PBNBH-MCPE) (Fig. 2B, curve a) and PBNBH-TNMCPE (Fig. 2B, curve b).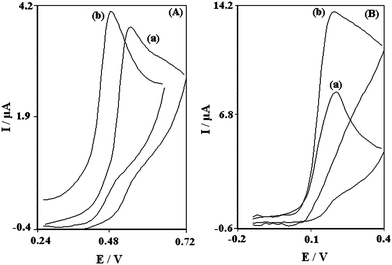 | ||
| Fig. 2 (A) Cyclic voltammograms of 0.6 mM DA in 0.1 M phosphate buffer solution (pH 7.0) at (a) Bare-CPE and (b) TNCPE. (B) Cyclic voltammograms of 0.6 mM DA in 0.1M phosphate buffer solution (pH 7.0) at (a) PBNBH-MCPE and (b) PBNBH-TNMCPE. In all cases the scan rate is 20 mV s−1. | ||
As shown, the anodic peak potential for DA oxidation at the PBNBH-MCPE (Fig. 2B, curve a) and PBNBH-TNMCPE (Fig. 2B, curve b) was about 190 mV, while at the TNCPE (Fig. 2A, curve b), the peak potential was about 480 mV. At the bare-CPE (Fig. 2A, curve a), the peak potential of DA was about 550 mV.
From these results, it was concluded that the best electrocatalytic effect for DA oxidation was observed at the PBNBH-TNMCPE (Fig. 2B, curve b). For example, results show that the peak potential of DA oxidation at the PBNBH-TNMCPE (Fig. 2B, curve b) shifted by about 290 and 360 mV toward negative values when compared with that at the TNCPE (Fig. 2A, curve b) and bare-CPE (Fig. 2A, curve a) respectively. Similarly, when comparing the oxidation of DA at the PBNBH-MCPE (Fig. 2B, curve a) and PBNBH-TNMCPE (Fig. 2B, curve b), a dramatic enhancement of the anodic peak current at the PBNBH-TNMCPE (Fig. 2B, curve b) relative to that obtained at the PBNBH-MCPE (Fig. 2B, curve a) was observed. In other words, the data clearly show that the combination of TiO2 nanoparticles and mediator (PBNBH) definitely improve the characteristics of DA oxidation.
Results of electrocatalytic oxidation of DA at the surface of chemically modified electrodes by other mediators are given in Table 1.32–34 Compared with other electrodes, the proposed electrode exhibits good electrocatalytic effect for DA oxidation.
| Substrate | Modifier | Method | pH | Peak potential shift/mV | Limit of detection/M | Dynamic range/M | Reference |
|---|---|---|---|---|---|---|---|
| Glassy Carbon Electrode | Cobalt hexa cyanoferrate | Voltammetry | 7.0 | — | 1.0 × 10−6 | 3 × 10−4–5 × 10−3 | 32 |
| Carbon Paste Electrode | Polypyrrole/ferrocyanide | Voltammetry | 7.0 | 95 | 1.0 × 10−5 | 2.0 × 10−4–95 × 10−4 | 33 |
| Carbon Paste Electrode | Cobalt salophe/tetraocthyl ammonium bromide | Voltammetry | 5.0 | 100 | 5.0 × 10−7 | 1.0 × 10−6–1.0 × 10−4 | 34 |
| Carbon Paste Electrode and TiO2 nanoparticles | 2,2′-[1,3 propanediylbis(nitriloethylidyne)]bis-hydroquinone | Voltammetry | 7.0 | 360 | 4.7 × 10−7 | 2.0 × 10−6–1.0 × 10−3 | This work |
In order to obtain information about the catalytic mechanism the cyclic voltammograms of DA solution at different scan rates were recorded. Fig. 3 shows the cyclic voltammograms of a PBNBH-TNMCPE at various scan rates obtained in 0.1 M phosphate buffer solution (pH 7.0) containing 0.5 mM DA. The anodic oxidation current of DA is proportional to the square root of the scan rate (Fig. 3A) indicating that at sufficiently positive potential the reaction is controlled by DA diffusion according to the following equation:31
| Ip = 3.01 × 105n[(1 − α)nα]1/2ACbD1/2υ1/2 | (6) |
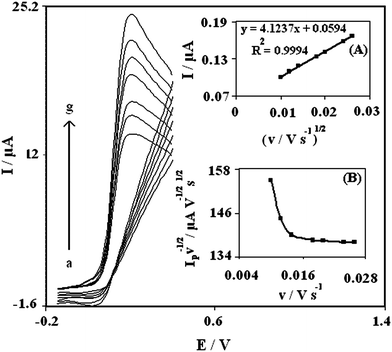 | ||
| Fig. 3 Cyclic voltammograms of 0.5 mM DA at PBNBH-TNMCPE at different scan rate in 0.1 M phosphate buffer solution (pH 7.0). (from a–g) 10, 12, 14, 18, 20, 24 and 26 mV s−1, respectively. Insets: (A) Variation of the electrocatalytic currents versus the square root of scan rate. (B) Variation of the normalized current (Ip/υ1/2) with scan rate. | ||
A plot of the normalized current (Ip/υ1/2) versus sweep rate shows the characteristic shape typical of an EC′ mechanism (Fig. 3B).
Andrieux and Saveant35 developed a theoretical model for such a mechanism and derived a relation between the peak current and the concentration of the substrate for the case of a slow rate, υ and a large catalytic rate constant, (k′h):
| Icat = 0.496nFACυ1/2(nFD/RT)1/2 | (7) |
Based on extensive computations, a working curve showing the relationship between numerical values of the constant, Icat/nFACb(DnFυ/RT)1/2, and logk′h/(DnFυ/RT)1/2] (Fig. 1a. of Andrieux's article) is given. Here D and Cb are the diffusion coefficient (cm2 s−1) and the bulk concentration (mol cm−3) of DA, respectively, and other parameters have their usual meanings. The value of catalytic reaction rate constant between DA and PBNBH-TNMCPE (k′h) can be calculated from such a working curve. For low scan rates (10–26 mV s−1), at the PBNBH-TNMCPE, an analysis of the CVs has given the average value for the ratio, Icat/nFACb(DnFυ/RT)1/2 to be 0.3, in the presence of 0.5 mM DA. Using this value and the theoretical paper by Andrieux and Saveant,35 the average values of k′h was found to be 2.31 × 10−3 cm s−1 (for the scan rates 6, 8, 10 mV s−1). This value of k′h explains well the sharp feature of the catalytic peak observed for catalytic oxidation of DA at the MTNCPE.
Fig. 4 shows linear sweep voltamograms of 0.5 mM DA in 0.1 M phosphate buffer solution (pH 7.0). The inset of Fig. 4 shows a Tafel plot that was drawn from data of the rising part of the current–voltage curve recorded at a scan rate of 25 mV s−1. This part of the voltammogram, known as the Tafel region, is affected by electron transfer kinetics between substrate (DA) and surface confined PBNBH-TNMCPE, assuming the deprotonation of substrate as a sufficiently fast step. In this condition, the number of electrons involved in the rate determining step can be estimated from the slope of the Tafel plot. A slope 0.0928 V decade−1 is obtained indicating a one electron transfer to be rate limiting assuming a transfer coefficient of (α = 0.36).
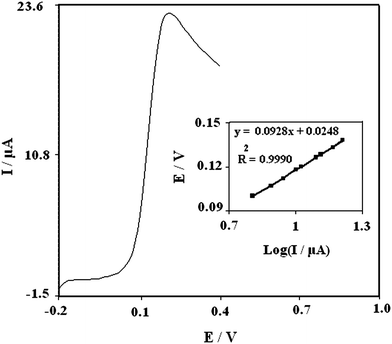 | ||
| Fig. 4 Linear sweep voltamogram of 0.5 mM DA at PBNBH-TNMCPE in 0.1 M phosphate buffer solution (pH 7.0). Inset shows the Tafel plot derived from the current potential curve recorded at a scan rate of 25 mV s−1 and the dependence of the peak potential, E to the log (I). | ||
3.4. Chronoamperometric studies
In chronoamperometry studies, the current, i, for the electrochemical reaction (at a mass-transport-limited rate) of an electroactive material (DA in this case) that diffuses to an electrode surface with a diffusion coefficient D is described by the Cottrell equation:31| I = nFAD1/2Cbπ −1/2t −1/2 | (8) |
3.5. Calibration plot and limit of detection
Since square wave voltammetry (SWV) has a much higher current sensitivity and better resolution than cyclic voltammetry, it was used to estimate the lower limit of detection of DA. In addition, the charging current contribution to the background current, which is a limiting factor in the analytical determination, is negligible in SWV mode. Voltammograms clearly show that the plot of peak current versus DA concentration is constituted of two linear segments with different slopes (slope: 0.428 μA μM−1 for first linear segment and 0.0399 μA μM−1 for second linear segment), corresponding to two different ranges of substrate concentration, 2.0–10.0 μM for first linear segment and 10.0–1000.0 μM for second linear segment. The decrease of sensitivity (slope) in the second linear range is likely to be due to kinetic limitation. The detection limit (2σ) for DA in the lower range region was found to be 0.47 μM. This value is comparable with values reported by other research groups (Table 1).3.6. Simultaneously determination of DA, UA and Trp
One of the main objectives of the present study was to develop a modified electrode with the capability of separating the electrochemical responses of DA, UA and Trp.The utilization of the PBNBH-TNMCPE for the simultaneous determination of DA, UA and Trp was demonstrated by simultaneously changing the concentrations of DA, UA and Trp. The SW voltammetric results show that the simultaneous determination of DA, UA and Trp with three well-defined anodic peaks is possible at the modified electrode (Fig. 5).
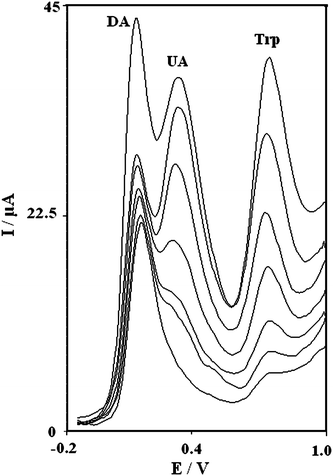 | ||
| Fig. 5 Square wave voltammograms of the MTNCPE in 0.1 M phosphate buffer solution (pH 7.0) in mixed solutions 2.0, 4.0, 8.0, 14.0, 16.0, 22.0, and 46.0 μM (DA), 10.0, 20.0, 40.0, 60.0, 100.0, 140.0 and 160.0 μM (UA) and 20.0, 70.0, 110.0, 210.0, and 280.0, 390.0, 500.0 μM (Trp) in potential scan rate 25 mV s−1. | ||
The sensitivities of the modified electrode towards the oxidation of DA, UA and Trp were found to be 0.4091 μA μM−1 (inset A of Fig. 6), 0.167 μA μM−1 (inset B of Fig. 6) and 0.0704 μA μM−1 (inset C of Fig. 6) respectively, whereas the sensitivities towards DA in the absence and presence of UA and Trp were found to be 0.4280 (absence of UA and Trp) and 04091 (presence of UA and Trp) μA μM−1. These results indicate that the oxidation processes of DA, UA and Trp at the PBNBH-TNMCPE are independent and therefore simultaneous measurements of the three analytes are possible without any interference. If the DA signal is affected by the UA and Trp signals, the above-mentioned slopes would be different.
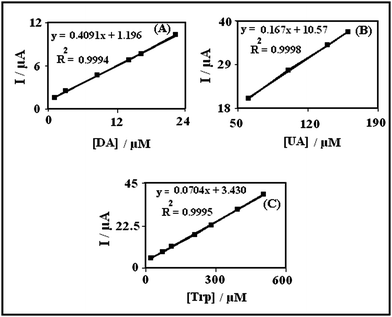 | ||
| Fig. 6 Plots of the peak currents as a function of concentration of: (A) DA with concentrations from 2.0–22.0 μM, (B) UA with concentrations from 60.0–160.0 μM and (C) Trp with concentrations from 20.0–500.0 μM in 0.1 M phosphate buffer solution (pH 7.0) at PBNBH-TNMCPE. | ||
3.7. The repeatability and stability of PBNBH-TNMCPE
The electrode capability for the generation of a reproducible surface was examined by cyclic voltammetric data (such as: Epa, Epc, Ipa and Ipc) obtained in optimum solution pH from four separately prepared PBNBH-TNMCPEs. The calculated RSD for various parameters accepted as the criteria for a satisfactory surface reproducibility was 1–4%.In addition, the long term stability of the PBNBH-TNMCPE was tested over a four-week period. Cyclic voltammetry of DA at the surface of PBNBH-TNMCPE after the modified electrode was stored in an atmosphere at room temperature shows the oxidation peak potential of DA was unchanged and the anodic peak current only decreased less than 2.2% of the initial oxidation peak current. The antifouling properties of modified electrode toward DA and its oxidation product were investigated by recording the cyclic voltammograms of this modified electrode before and after using in the presence of DA.
The cyclic voltammetry of DA at the surface of PBNBH-TNMCPE after 12 repetition cycles at a scan rate 25 mV s−1 shows the oxidation peak potential of DA was not changed and the anodic peak current was decreased by less than 3.1%. However we regenerated the surface of PBNBH-TNMCPE before each experiment.
3.8. Determination of DA in real samples
The proposed PBNBH-TNMCPE was found to work well under laboratory conditions. The electrode was also successfully applied to the direct determination of DA content of DA ampoule. The DA content in DA ampoule was determined by the standard addition method in order to prevent of any matrix effect. The results are presented in Table 2.| No | Initial concentration (×10−4 M) | DA added (×10−4 M) | DA found (×10−4 M) | Recovery (%) | RSD (%) |
|---|---|---|---|---|---|
| 1 | 0.21 | 1.5 | 1.66 | 97 | 0.1 |
| 2 | 0.21 | 3.0 | 3.1 | 96.5 | 0.3 |
| 3 | 0.21 | 5.9 | 5.8 | 95 | 0.4 |
| 4 | 0.21 | 8.7 | 9.1 | 102 | 0.6 |
4. Conclusion
Simultaneous trace determination of DA, UA and Trp was achieved by using a carbon paste electrode with PBNBH-TNMCPE. Large peak separations between DA, UA and Trp allow the detection and determination of DA, UA and Trp simultaneously at MTNCPE using SWV. The catalytic peak current obtained from SWV was linearly dependent on the DA concentration in the range 2.0–1000.0 μM. The detection limit (2σ) for DA was 0.47 μM. The proposed method can be applied to the detection of DA in pharmaceutical samples.Acknowledgements
The authors would like to thank Yazd University Research Council, IUT Research Council and Excellence in Sensors for financial support of this research. We gratefully acknowledge Dr. N. Taghavinia of Sharif University of Technology for preparing the TiO2 nanoparticles.References
- P. Alivisatos, Nat. Biotechnol., 2004, 22, 47 CrossRef.
- M. H. Mashhadizadeh and H. Khani, Anal. Methods, 2010, 2, 24 RSC.
- K. Wu, J. Fei and S. Hu, Anal. Biochem., 2003, 318, 100 CrossRef CAS.
- J. W. Mo and B. Ogorevc, Anal. Chem., 2001, 73, 1196 CrossRef CAS.
- Y. Liu, J. Huang, H. Hou and T. You, Electrochem. Commun., 2008, 10, 1413.
- A. Salimi, H. Mamkhezri and R. Hallaj, Talanta, 2006, 70, 823 CrossRef CAS.
- M. Mazloum-Ardakani, Z. Taleat, H. Beitollahi and H. Naeimi, Anal. Methods, 2010, 2, 149 RSC.
- S. H. Huang, Y. C. Shih, C. Y. Wu and C. J. Yuan, Biosens. Bioelectron., 2004, 19, 1627 CrossRef CAS.
- E. Miland and A. J. Miranda Ordieres, Talanta, 1996, 43, 785 CrossRef CAS.
- L. Zhang and X. Lin, Analyst, 2001, 126, 367 RSC.
- R. P. Baldwin and K. N. Thomsen, Talanta, 1991, 38, 1 CrossRef CAS.
- X. Jiang and X. Lin, Analyst, 2005, 130, 391 RSC.
- M. Mazloum-Ardakani, H. Beitollahi, B. Ganjipour, H. Naeimi and M. Nejati, Bioelectrochemistry, 2009, 75, 1 CrossRef CAS.
- K. Horwitt, C. C. Harvey, W. S. Rothwell, J. L. Cutler and D. Haffron, J. Nutr., 1965, 60, 1.
- A. Coppen, Br. J. Psychiatry, 1967, 113, 1237 Search PubMed.
- R. J. Reiter, Exp. Gerontol., 1995, 30, 199 CrossRef CAS.
- G. Biggio, F. Fadda, P. Fanni, A. Tagliamonte and G. L. Gessa, Life Sci., 1974, 14, 1321 CrossRef CAS.
- V. Simonneaux and C. Ribelayga, Pharmacol. Rev., 2003, 55, 325 CrossRef CAS.
- A. Babaei, M. Zendehdel, B. Khalilzadeh and A. Taheri, Colloids Surf., B, 2008, 66, 226 CrossRef CAS.
- Z. Chen, K. Okamura, M. Nanaki and T. Nagaoka, Anal. Sci., 2002, 18, 417 CrossRef CAS.
- A. A. Ensafi and R. Hajian, Anal. Chim. Acta, 2006, 580, 236 CrossRef CAS.
- A. M. Krstulovic, M. J. Friedman, H. Colin, G. Guiochon and K. Pajer, J. Chromatogr., A, 1984, 297, 271 CrossRef CAS.
- Z. Shen, Z. Sun, L. Wu, K. Wu, S. Sun and Z. Huang, J. Chromatogr., A, 2002, 979, 227 CrossRef CAS.
- M. Mazloum-Ardakani, H. Beitollahi, M. A. Sheikh Mohseni, A. Benvidi, H. Naeimi, M. Nejati-Barzoki and N. Taghavinia, Colloids Surf., B, 2010, 76, 82 CrossRef CAS.
- H. Beitollahi, M. Mazloum Ardakani, B. Ganjipour and H. Naeimi, Biosens. Bioelectron., 2008, 24, 362 CrossRef CAS.
- H. L. Zhang, Y. Liu, G. S. Lai, A. M. Yu, Y. M. Huang and C. M. Jin, Analyst, 2009, 134, 2141 RSC.
- A. D. Namor and I. Abbas, Anal. Methods, 2010, 2, 63 RSC.
- M. Mazloum Ardakani, Z. Taleat, H. Beitollahi, M. Salavati-Niasari, B. B. F. Mirjalili and N. Taghavinia, J. Electroanal. Chem., 2008, 624, 73 CrossRef CAS.
- E. Laviron, J. Electroanal. Chem., 1979, 101, 19 CrossRef CAS.
- M. Sharp, M. Petersson and K. Edstrom, J. Electroanal. Chem., 1979, 95, 123 CrossRef CAS.
- A. J. Bard and L. R. Faulkner. Electrochemical methods. Fundamentals and applications, 2nd ed. Wiley, New York 2001 Search PubMed.
- Z. Xun, C. Cai, W. Xing and T. Lu, J. Electroanal. Chem., 2003, 545, 19 CrossRef CAS.
- J. B. Raoof, R. Ojani and S. Rashid-Nadimi, Electrochim. Acta, 2005, 50, 4694 CrossRef CAS.
- S. Shahrokhian and H. R. Zare-Mehrjardi, Sens. Actuators, B, 2007, 121, 530 CrossRef.
- C. P. Andrieux and J. M. Saveant, J. Electroanal. Chem., 1978, 93, 163 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |