DOI:
10.1039/C0AY00021C
(Paper)
Anal. Methods, 2010,
2, 492-498
Rapid determination of six kavalactones in kava root and rhizome samples using Fourier transform infrared spectroscopy and multivariate analysis in comparison with gas chromatography
Received
12th January 2010
, Accepted 24th February 2010
First published on
17th March 2010
Abstract
Kava has been used as a folk medicine and well-known traditional beverage. This study focused on quantitative analyses of kavalactones in kava samples using Fourier transform infrared spectroscopy (FTIR) integrated with an attenuated total reflectance (ATR) accessory and multivariate calibration analysis. The calibration set consisted of 50 standard mixtures which were made up of six major kavalactones. Derivative transformations (1st and 2nd), mathematical enhancements such as mean centering and variance scaling, and multivariate regression by partial least square (PLS) were implemented to develop and enhance the calibration model. Distinct peaks associated with the six kavalactones varied in the range between 1100 and 1800 cm−1. The PLS-2nd derivative calibration model based on the 50 standard mixtures with mean centering data processing showed standard errors of calibration (SEC) of 0.72–1.73, standard errors of prediction (SEP) of 0.98–3.52 and correlation coefficients (R2) of 0.81–0.98 for model validation. This model was used for the analysis of these kavalactones in kava samples and then compared with gas chromatography (GC). Results showed that the FTIR-predicted values were similar to GC-determined values (R2, 0.75–0.98), indicating that the FTIR method is suitable for determination of the contents of the six kavalactones in kava samples and thereby the chemotypes.
Introduction
Piper methysticum G. Forst, popularly known as kava, is a plant species widely distributed throughout the south Pacific region such as Vanuatu, Fiji, Samoa, Tonga, Micronesia, Hawaii, and Wallis-Futuna.1 The kava beverage and other preparations from kava rhizome and roots have been used for thousands of years in these regions.1 Historic use indicates that kava is safe and effective in the treatment of anxiety, restlessness, and insomnia. Kava is particularly capable of bringing a state of relaxation without loss of mental alertness.2,3 Many chemical components are present in Piper methysticum, for example, kavapyrones, alkaloids, steroids, chalcones, long-chain fatty acids and alcohols.4,5 Eighteen different kavalactones have been found in rhizome and root extracts of kava; desmethoxyyangonin (DMY), yangonin (Y), dihydrokavain (DHK), kavain (K), dihydromethysticin (DHM), and methysticin (M) (Fig. 1) are among the most abundant kavalactones.4,6,7 These six major kavalactones are used to define a particular kava sample's chemotype8 and assigned with numbers based on the sequence of elution on high performance liquid chromatography (HPLC): 1 = DMY; 2 = DHK; 3 = Y; 4 = K; 5 = DHM; and 6 = M. Chemotypes can be identified by listing the numbers of the corresponding kavalactones in decreasing order of their proportion in the sample. This method of chemotype coding allows the indication of a “signature” of the raw material and the comparison between kavalactone composition and effect experienced on ingestion.8
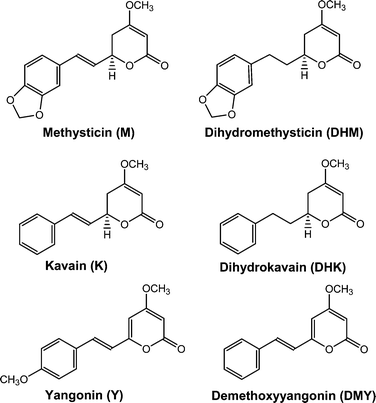 |
| | Fig. 1 Molecular structures of the six major kavalactones. | |
The content of kavalactones in kava roots and rhizome is related to the cultivars, geographic location of the cultivation, and the age and different parts of the plant. The relative proportion of the six major kavalactones in kava extracts impact the pharmacological effects and intensity and, therefore, it is important to determine the content and chemotypes of kavalactones.1 Analytical methods are necessary for quality control of kava materials and beverages. Many analytical techniques are used for the analysis of kavalactones and quality control for standardized phytotherapeutical and nutraceutical preparations.6–10 Kavalactones are typically extracted with organic solvents under atmospheric or pressurized conditions.11 Kavalactones in the extracts have been primarily resolved and detected with HPLC and/or liquid chromatography-mass spectrometry (LC-MS),6,12,13 gas chromatography (GC),14,15 thin-layer chromatography,16 and capillary zone electrophoresis.17 Near-infrared reflectance (NIR) spectroscopy and multivariate calibration analysis have been recently studied for measurement of the major kavalactones in kava plants.9,10,18
Each method has disadvantages as well. For example, two major kavalactones, methysticin and yangonin, are often not well resolved by GC and the former is degraded in the GC injection port due to its poor thermo-stability.19 HPLC methods are less sensitive and the use of an internal standard is advisable to minimize deviations. It also requires use of toxic and volatile organic solvents. Furthermore, sample preparation for normal-phase column chromatography is generally complicated by the fact that the injection sample must be free of all traces of water. Reversed-phase HPLC provides good separation but the analysis time is long. GC-MS is also a powerful analytical tool because GC offers high separation and MS gives information about constituent structures; however, the high temperature of the injection port can cause the decomposition of methysticin.15 In addition, supercritical fluid chromatography and micellar electrokinetic chromatography have been proposed for the analysis of kavalactones. However, they are too preliminary to be adopted as alternative methods.19
Fourier transform infrared (FTIR) spectroscopy with smart accessory has advantages of non-destruction, high speed, high sensitivity, and simple operation. Combined with multivariate calibration analysis, the FTIR method has been successfully used in many areas, in particular for food analyses.20–28 The objective of this study was to develop a reliable FTIR method for quantifying the six major kavalactones to estimate kava chemotypes. The first step was intended to develop a calibration model of a partial least square (PLS) regression based on 50 standard mixtures of the six kavalactones, integrated with different derivative transformations (1st and 2nd). The model predictability was then validated with 10 mixtures of the six kavalactones that were unused for model development. Finally, the model was validated in comparison with a GC method.
Experimental
Kava sample extracts and preparation of standard mixtures of kavalactones
Root and rhizome samples of two cultivars were obtained from Waimanalo farm in Hawaii. The kavalactones DMY, DHK, Y, K, DHM, and M were purchased from Sigma-Aldrich Corp. (St. Louis, MO, USA). All kava samples were lyophilized and well ground. Each sample (0.1 g) was extracted with 10 ml of hexane, acetone, methanol, ethyl acetate, ethanol, or dimethylsulfoxide (DMSO) and sonicated for 30 min. After filtration, the solvent was replaced with DMSO under a gentle stream of high purity nitrogen gas followed by FTIR analyses. It is known that different organic solvents have various degrees of extraction efficiencies for different kavalactones.29 Kava extracts produced by using the six different extracting solvents would contain varying profiles of kavalactones, which would be equivalents to different kava samples and, thus, overcome the limitation of the few kava samples for the FTIR method validation. A total of 24 kava sample extracts were produced for this study (root and rhizome samples from these two very different cultivars with 6 extracting solvents). The kava extracts from using various solvents were expected to challenge and confirm most of potential optical interferences to occur during the FTIR measurement.
According to the previous studies,9,10 the calibration sets were comprised of 50 standard mixtures of kavalactones with known concentrations of DMY (18–136 mg l−1), DHK (5–266 mg l−1), Y (22–134 mg l−1), K (6–271 mg l−1), DHM (24–213 mg l−1), and M (11–99 mg l−1) (Table 1). DMSO was used to dissolve the kavalactones. In addition, 10 solutions containing standard mixtures of six kavalactones were separately prepared for model validation.
Table 1 Calibration sets (mg l−1) of 50 standard mixtures in DMSO that were made up of six major kavalactones (The ratios of concentrations of the six kavalactones were adapted from Lasme et al.9 and Gautz et al.10)
| Solution Number |
DMY |
DHK |
Y |
K |
DHM |
M |
Solution Number |
DMY |
DHK |
Y |
K |
DHM |
M |
| 1 |
35.5 |
74.5 |
49.5 |
135.0 |
31.5 |
21.5 |
26 |
44.5 |
170.5 |
116.00 |
153.0 |
124.0 |
98.5 |
| 2 |
45.5 |
110.5 |
78.5 |
202.5 |
35.5 |
32.0 |
27 |
36.5 |
183.0 |
98.00 |
134.0 |
112.0 |
74.5 |
| 3 |
43.5 |
148.5 |
121.0 |
236.0 |
52.5 |
44.0 |
28 |
35.5 |
135.0 |
83.00 |
123.0 |
89.0 |
71.0 |
| 4 |
38.5 |
99.0 |
102.5 |
169.0 |
54.0 |
40.0 |
29 |
28.0 |
86.5 |
51.00 |
85.0 |
53.5 |
48.0 |
| 5 |
40.0 |
123.0 |
89.0 |
187.5 |
52.0 |
46.5 |
30 |
30.5 |
131.5 |
67.50 |
94.5 |
88.5 |
57.5 |
| 6 |
49.5 |
153.0 |
133.5 |
235.5 |
74.5 |
52.0 |
31 |
40.0 |
151.5 |
99.00 |
135.0 |
103.5 |
73.0 |
| 7 |
58.0 |
172.0 |
132.5 |
270.5 |
80.5 |
62.0 |
32 |
38.5 |
148.5 |
97.00 |
132.0 |
100.0 |
70.5 |
| 8 |
37.0 |
116.5 |
103.0 |
163.0 |
47.5 |
35.5 |
33 |
32.5 |
128.5 |
74.50 |
106.0 |
106.5 |
61.0 |
| 9 |
42.0 |
108.0 |
97.0 |
176.5 |
48.0 |
43.0 |
34 |
43.5 |
155.5 |
96.50 |
135.0 |
126.5 |
90.0 |
| 10 |
35.0 |
114.5 |
94.0 |
160.5 |
48.0 |
39.5 |
35 |
29.5 |
111.5 |
57.00 |
91.5 |
88.5 |
69.5 |
| 11 |
25.5 |
102.5 |
87.0 |
137.0 |
47.5 |
34.5 |
36 |
28.5 |
97.5 |
71.50 |
87.0 |
85.5 |
69.5 |
| 12 |
33.5 |
97.0 |
84.5 |
144.0 |
41.0 |
35.5 |
37 |
20.5 |
112.0 |
64.50 |
84.5 |
76.0 |
65.0 |
| 13 |
30.5 |
72.0 |
67.0 |
121.0 |
37.0 |
34.5 |
38 |
24.0 |
106.0 |
65.50 |
81.0 |
87.0 |
72.5 |
| 14 |
39.0 |
108.5 |
95.5 |
186.5 |
59.5 |
56.5 |
39 |
28.5 |
103.0 |
55.00 |
95.5 |
96.5 |
73.0 |
| 15 |
19.0 |
56.0 |
37.0 |
96.0 |
33.0 |
31.5 |
40 |
21.0 |
136.5 |
72.50 |
100.5 |
82.5 |
66.5 |
| 16 |
23.5 |
74.0 |
44.0 |
117.5 |
36.0 |
33.5 |
41 |
45.5 |
260.5 |
93.00 |
130.0 |
154.5 |
65.5 |
| 17 |
23.0 |
58.5 |
42.0 |
119.0 |
24.0 |
33.5 |
42 |
27.0 |
188.0 |
89.50 |
98.0 |
170.5 |
79.0 |
| 18 |
21.5 |
62.0 |
40.0 |
100.5 |
34.5 |
33.0 |
43 |
18.0 |
108.5 |
62.00 |
80.0 |
83.0 |
69.0 |
| 19 |
22.5 |
73.5 |
35.0 |
111.5 |
28.5 |
30.0 |
44 |
29.5 |
22.0 |
73.50 |
43.5 |
86.0 |
60.5 |
| 20 |
32.0 |
75.0 |
71.5 |
134.5 |
37.5 |
37.5 |
45 |
35.0 |
26.5 |
22.00 |
14.0 |
65.5 |
12.0 |
| 21 |
39.0 |
142.0 |
88.0 |
146.5 |
91.5 |
91.0 |
46 |
135.5 |
128.5 |
57.50 |
23.5 |
212.5 |
29.5 |
| 22 |
39.0 |
91.5 |
92.0 |
163.5 |
43.0 |
36.5 |
47 |
55.5 |
24.0 |
31.00 |
7.5 |
105.0 |
11.0 |
| 23 |
47.0 |
265.5 |
64.0 |
193.0 |
36.0 |
35.0 |
48 |
118.0 |
139.0 |
42.00 |
17.0 |
156.0 |
25.5 |
| 24 |
31.5 |
123.0 |
74.5 |
96.5 |
85.5 |
48.0 |
49 |
102.0 |
106.0 |
36.50 |
15.0 |
143.5 |
16.5 |
| 25 |
42.0 |
202.0 |
81.0 |
132.5 |
122.0 |
66.0 |
50 |
40.5 |
5.0 |
22.50 |
6.5 |
99.0 |
18.5 |
FTIR measurement
FTIR measurements were performed on a Nicolet 6700 spectrometer (Thermo Electron Corp., Madison, WI, USA) equipped with a deuterated triglycine sulfate detector and smart attenuated total reflectance kit (ARK) that was an advanced multi-bounce horizontal attenuated reflectance (HATR) accessory. The HATR accessory was comprised of a ZnSe crystal sample cell with a slit angle of 45° and refractive index of 2.4 at 1000 cm−1, providing high energy throughput and 12 reflections with a penetration depth (infrared beam) of 2.0 μm.
Synthetic mixtures of kavalactones and kava sample extracts were scanned at room temperature (ca 25 °C). Single beam spectra (4000–400 cm−1) of standard mixtures of kavalactones and the absorbance spectra of kava extracts in DMSO were obtained against DMSO background at a resolution of 8 cm−1 and a total of 256 co-added scans. The spectra were collected and recorded with OMNIC software (Thermo Electron). The ATR crystal was carefully cleaned with pure ethanol to remove any residues on surface of the HATR cell. The data were averaged from duplicate runs by GRAMS software (Version 8.0, Thermo Fisher Scientific, Inc., Woburn, MA, USA) prior to multivariate analysis. Kava extracts were analyzed both immediately and 30 min after contact with the ATR crystal. The two spectra showed no difference. The FTIR spectral collection time for each measurement was less than 4 min. The total time for analysis of one sample including sample preparation was less than 10 min.
Data analysis
The derivative transformations of spectral data (1st and 2nd) and mathematical enhancer for multivariate analysis such as mean centering, and variance scaling were used to develop a calibration model for accurate prediction of the concentrations of the six kavalactones. Mean centering and variance scaling are normally applied to mathematically enhance the important sample-to-sample differences by removing redundant information.30,31 Mean centering involves calculation of the average values of all spectra and subsequent subtraction of them from each spectrum. Variance scaling emphasizing small variations in the data is calculated by dividing the response at each spectral data point by the standard deviation of the responses of all training spectra at that point. Derivative transformations can partially compensate for baseline offset between samples and reduce instrument drift effects.32,33 The predictability of the models was tested by computing the standard error of calibration (SEC) for calibration set and the standard error of prediction (SEP) for validation data sets as below:25| | 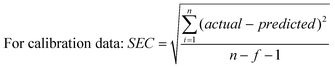 | (1) |
| | 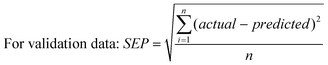 | (2) |
The term “actual” refers to the kavalactone content, and “predicted” refers to the content computed by PLS of the sample spectra; n is the number of samples in the calibration set, and f refers to the number of factors in the calibration model.
The undesirable variations in the spectra were removed using different derivative transformations and mathematical enhancements given by the GRAMs software.32,34 The prediction intervals at a 95% confidence were computed for each regression model (in both calibration and external prediction) in order to estimate and compare the model reliability. The discriminant PLS analysis followed by canonical variate analysis (CVA) was performed by using WinDAS software (Wiley and Sons, Chichester, UK) to differentiate various kavalactones. The PLS calibrations obtained were tested by “split sample” cross validation, in which successive groups of widely separated observations were held out as the test set. The number of factors chosen is usually the one that minimizes the predicted residual sum of squares (PRESS) with the highest R2 value, which should be as high as possible.34,35
GC analysis
The standard and sample solutions were analyzed on a Hewlett Packard (HP) 5890 gas chromatograph-flame ionization detector (Palo Alto, CA, USA). The GC column was a 30 m HP-5MS capillary column (30 m × 0.25 mm, 0.25 μm; J&W Scientific). The flow rate of helium carrier gas was maintained at 1 ml min−1. The column temperature was started from 100 °C, raised to 260 °C at a rate of 30 °C min−1, and then held at 260 °C for 30 min. All samples (1 μl) were injected in splitless mode with an HP autosampler. Fig. 2 shows GC chromatograms of six kavalactone standards and a typical kava extract.
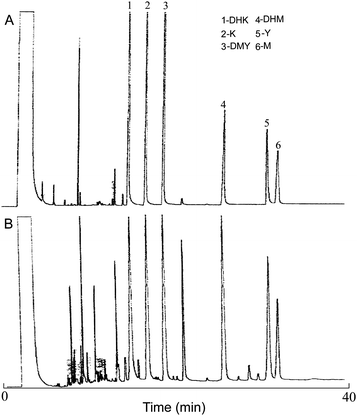 |
| | Fig. 2 GC chromatograms of six kavalactone standards (A) and a typical kava extract (B). | |
Quality control and quality assurance
Sample preparations and analytical procedures were performed according to quality assurance and quality control measures. Limits of detection (LODs) were derived from the blanks and quantified as within three standard deviations of the mean blank intensity based on IR absorbance peaks of main chemical groups in the six major kavalactones between 1100 and 1800 cm−1. Significant IR absorbance peaks specific to the six kavalactones were found in the range between 1100 and 1800 cm−1 even at concentrations as low as 2 mg l−1. LODs were estimated by testing the six kavalactone standards at concentrations from 5 to 10 mg l−1.
Results and discussion
Characteristics of IR spectra of the six kavalactones and selection of optimal spectral range for qualitative and quantitative analysis
A good IR solvent needs to meet several criteria. It should have minimum absorbance at the informative fingerprint region that will be used for qualitative and quantitative analysis, low volatility that allows convenient handling of the samples and measurement, and inertness that does not react with the analytes or components in the matrices. DMSO can meet these criteria and was selected as the solvent in this study. Fig. 3 shows representative IR spectra of DMSO and the standard kavalactones desmethoxyyangonin (DMY), dihydrokavain (DHK), yangonin (Y), kavain (K), dihydromethysticin (DHM), and methysticin (M). The spectral region between 1100 cm−1 and 1800 cm−1 contained unique absorption bands that would be sufficient for discrimination of these kavalactones and, thus, was selected for design of calibration model. The peaks of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CC stretching are in between 1600 cm−1 and 1800 cm−1. Absorption bands in the region between 1500 and 1100 cm−1 are mainly bending vibrations of O–C–H, C–C–H, C–O–C and C–O–H. Absorption bands of DMY include 1723 (CO), 1640 (CC), 1566 and 1252 (C–O–C) cm−1. The spectrum of DHK includes bands at 1707 (CO), 1624, and 1225 cm−1. Yangonin shows bands at 1717 (CO), 1644 (CC), 1603, 1555, and 1256 cm−1. Kavain has absorption bands 1717 (CO), 1626 (CC), and 1248 cm−1. Absorption bands of DHM include 1721 (CO), 1644, 1500, and 1256 cm−1. Absorption bands of M include 1711 (CO), 1628 (CC), and 1252 cm−1.36 In addition, the selected IR spectral range of 1100–1800 cm−1 of the six kavalactones includes many small but distinguishable absorbance peaks (not displaying in Fig. 3). Fig. 4A1 and 4A2 show raw and 2nd-derivative spectra of 50 standard mixtures of the six kavalactones used for design of the calibration model, respectively. Fig. 4B1 and 4B2 show raw and 2nd-derivative spectra of 24 kava sample extracts, respectively, of which the absorbance peaks in the selected region were similar to those of standard mixtures of the kavalactones.
O and CC stretching are in between 1600 cm−1 and 1800 cm−1. Absorption bands in the region between 1500 and 1100 cm−1 are mainly bending vibrations of O–C–H, C–C–H, C–O–C and C–O–H. Absorption bands of DMY include 1723 (CO), 1640 (CC), 1566 and 1252 (C–O–C) cm−1. The spectrum of DHK includes bands at 1707 (CO), 1624, and 1225 cm−1. Yangonin shows bands at 1717 (CO), 1644 (CC), 1603, 1555, and 1256 cm−1. Kavain has absorption bands 1717 (CO), 1626 (CC), and 1248 cm−1. Absorption bands of DHM include 1721 (CO), 1644, 1500, and 1256 cm−1. Absorption bands of M include 1711 (CO), 1628 (CC), and 1252 cm−1.36 In addition, the selected IR spectral range of 1100–1800 cm−1 of the six kavalactones includes many small but distinguishable absorbance peaks (not displaying in Fig. 3). Fig. 4A1 and 4A2 show raw and 2nd-derivative spectra of 50 standard mixtures of the six kavalactones used for design of the calibration model, respectively. Fig. 4B1 and 4B2 show raw and 2nd-derivative spectra of 24 kava sample extracts, respectively, of which the absorbance peaks in the selected region were similar to those of standard mixtures of the kavalactones.
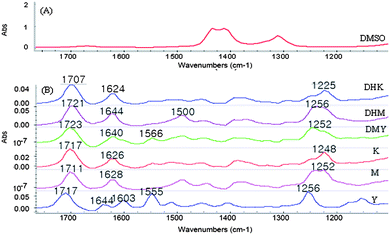 |
| | Fig. 3 FTIR spectra of DMSO (A) and six individual kavalactones (B). Note: The absorbance of DMSO was subtracted from the spectra of the kavalactones. The spectrum of DMSO shows weak absorbance bands in the range of 1100–1800 cm−1. | |
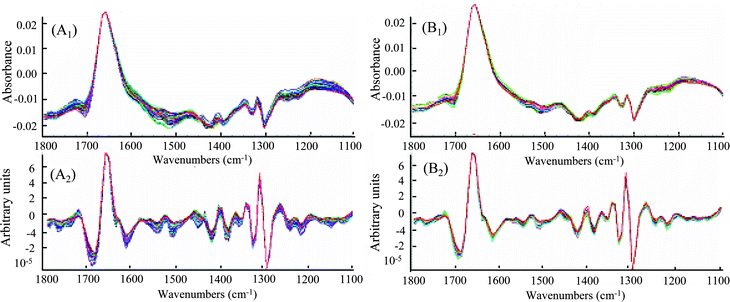 |
| | Fig. 4 FTIR spectra of 50 standard mixtures of six major kavalactones. (A1): calibration sets, (A2): 2nd-derivation of (A1), (B1): kava root and rhizome extracts and (B2): 2nd-derivation of (B1). | |
Discriminant analysis of the six kavalactones
When discriminant analysis was applied to interpret the FTIR spectra of different kavalactones, canonical variate (CV) scores could be used as a means to maximize inter-group variances and minimize intra-group variances. All the spectral data were selected from the range of 1100–1800 cm−1 of the FTIR spectra and transformed with the secondary derivation. Fig. 5 shows a bidimensional representation of the two CV scores for the six kavalactones based on PLS data compression with a 95% confidence. The smaller the distance between the groups is, the greater the similarity can be found. DHK, DHM and K were overlapped with high similarity, particularly DHK and DHM (Fig. 5). DMY was separated well from DHK and DHM, but was slightly overlapped with K. M and Y were clearly distant from any others, suggesting that those two can be differentiated from the rest of them with high distinction.
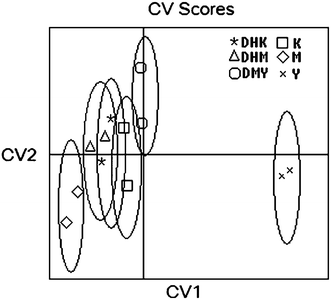 |
| | Fig. 5 Canonical variate (CV) plot of the six kavalactones discriminated by the developed FTIR-PLS calibration model. | |
Developing PLS regression model for quantitative analysis of the six kavalactones in kava rhizome and root samples
The SEC for calibration set and SEP for validation data were used to evaluate the accuracy of the model prediction. Results showed that the PLS calibration model based on the 2nd-derivative spectra coupled with mean centering provided low SEC values (0.72–1.73) and high correlation coefficients (R2, 0.87–0.99) for calibration sets and low SEP values (0.98–3.52) and high correlation coefficients (R2, 0.84–0.98) for validation sets (Table 2). A comparison of the actual and predicted data of the validation set is shown in Table 3. The PLS calibration model could be applied for the accurate detection of the six kavalactones in kava sample extracts. The main advantage of the associated spectral transformation in the process of developing a PLS calibration model is that it can narrow several variables to one useful factor. Therefore, in the subsequent experiments, the PLS regression model based on 2nd-derivative transformations coupled with mean centering was applied to quantitative analysis of the six kavalactones in the kava sample extracts.
Table 2 Summary of calibration and validation statistical parameters of the FTIR-PLS calibration model using 2nd-derivative transformations coupled with mean centering data processing methods
| Kavalactones |
PLS factors |
Calibration |
Validation |
| R2 |
SEC |
R2 |
SEP |
| DMY |
3 |
0.88 |
1.46 |
0.84 |
0.98 |
| DHK |
3 |
0.95 |
1.73 |
0.92 |
2.07 |
| Y |
4 |
0.96 |
0.72 |
0.93 |
3.52 |
| K |
7 |
0.99 |
0.85 |
0.98 |
2.64 |
| DHM |
7 |
0.98 |
0.73 |
0.98 |
2.26 |
| M |
1 |
0.87 |
1.29 |
0.81 |
3.07 |
Table 3 Comparison of the actual values and FTIR-PLS predictions of the six kavalactones in ten validation sets (Unit: mg l−1)
| DMY |
DHK |
Y |
K |
DHM |
M |
| Actual |
PLSMC |
Actual |
PLSMC |
Actual |
PLSMC |
Actual |
PLSMC |
Actual |
PLSMC |
Actual |
PLSMC |
| 33.5 |
34.4 |
76.5 |
79.4 |
52.5 |
60.0 |
150.0 |
145.6 |
27.5 |
29.9 |
26.5 |
23.8 |
| 42.5 |
43.9 |
114.5 |
115.4 |
81.5 |
84.3 |
200.5 |
197.1 |
35.5 |
39.7 |
33.0 |
38.4 |
| 43.5 |
46.0 |
149.5 |
146.5 |
117.0 |
113.6 |
235.0 |
234.0 |
46.5 |
43.5 |
45.0 |
51.8 |
| 51.5 |
50.4 |
171.5 |
176.4 |
129.5 |
128.4 |
233.5 |
224.4 |
77.5 |
76.7 |
58.5 |
63.3 |
| 19.5 |
18.2 |
47.5 |
26.2 |
38.0 |
37.4 |
93.0 |
89.6 |
34.0 |
39.4 |
30.5 |
27.1 |
| 43.5 |
40.6 |
189.0 |
188.9 |
79.0 |
75.5 |
138.0 |
146.7 |
114.5 |
112.6 |
67.0 |
68.8 |
| 35.5 |
32.6 |
145.0 |
130.6 |
87.0 |
81.6 |
136.0 |
131.6 |
78.0 |
81.2 |
66.0 |
60.0 |
| 38.5 |
37.9 |
150.0 |
156.4 |
100.0 |
95.8 |
132.5 |
124.7 |
104.5 |
109.6 |
50.5 |
71.3 |
| 32.5 |
34.5 |
120.5 |
116.9 |
72.5 |
69.9 |
103.5 |
106.9 |
105.0 |
102.7 |
60.0 |
57.2 |
| 27.0 |
30.3 |
30.5 |
33.2 |
32.5 |
36.6 |
12.5 |
13.2 |
66.5 |
76.9 |
22.5 |
23.0 |
Quantification of the six kavalactones in kava sample extracts
The developed PLS-2nd derivative calibration model was used to estimate the content of the six kavalactones and thereby the chemotypes in the 24 extracts of the kava samples. Table 4 shows comparison of the chemotypes determined by GC and FTIR. In general, chemotype results are quite satisfactory for all extracts. The chemotypes of 10 out of the 24 kava extracts determined by GC and FTIR were identical and those of nine of the 24 extracts differed only by one neighboring position switch (e.g., 245361 by GC vs. 245631 by FTIR for kava extract 3). The chemotypes of extracts 7, 8, 15, 16, and 17 determined by the two methods differed in switches of two positions (e.g., 143265 by GC vs.416325 by FTIR for kava extract 7, 243165 by GC vs. 246315 by FTIR for kava extract 15). The FTIR-PLS predicted values of the six kavalactones were very similar to the GC-determined values. The R2 between the FTIR-PLS predicted and GC-determined values ranged from 0.75 to 0.98. This result indicates the potential of the PLS-2nd derivative calibration model to quantitatively measure the six kavalactones in kava samples. In addition to kavalactones, many solvent-extractable components are present in kava root and rhizome samples and probably have IR absorption in the selected spectral region (1100–1800 cm−1), which may result in deviation of the predicted results from the actual values, particularly methysticin (R2, 0.75) and yangonin (R2, 0.78) (Table 4). It is noteworthy that although the average GC and FTIR values were the same for methysticin (0.95%) and very similar for yangonin (1.10% by GC and 1.22% by FTIR), most GC-determined values of methysticin and yangonin were lower than the FTIR values (Table 4), which may be explained with poor stability of yangonin11 and easy thermo-degradation of methysticin19 and less background interferences relative to FTIR. The yangonin content in the kava extracts 17 and 20 determined by GC was 3.0–4.5-fold lower than that by FTIR (0.24% vs. 0.73% for extract 17 and 0.20% vs. 0.90% for extract 20, GC vs. FTIR). The methysticin content in the kava extract 20 measured by GC was 8.4-fold lower than that by FTIR (0.07% vs. 0.59%, GC vs. FTIR) while its content in the extract 24 measured by GC was 4.2-fold higher than that by FTIR (0.55% vs. 0.13%, GC vs. FTIR). These large disagreements have caused the low R2 values for methysticin and yangonin. The FTIR-PLS predicted values of the other four kavalactones agreed very well with the GC values (R2, 0.89–0.98).
Table 4 Comparison between the concentrations of kavalactones in the kava sample extracts determined by GC and FTIR (unit: %)
| Kava extractsa |
DMY |
DHK |
Y |
K |
DHM |
M |
Total KLb% |
Chemotype |
| GC |
FTIR |
GC |
FTIR |
GC |
FTIR |
GC |
FTIR |
GC |
FTIR |
GC |
FTIR |
GC |
FTIR |
GC |
FTIR |
|
Two root and two rhizome samples were extracted with six extracting solvents to generate a total of 24 kava sample extracts containing various profiles of the kavalactones as determined by GC. These 24 kava extracts were used to validate the FTIR-PLS-2nd-spectral derivative model coupled with mean centering data processing in comparison with GC determinations.
KL, kavalactones.
|
| 1 |
1.39 |
1.11 |
7.37 |
7.04 |
1.53 |
1.84 |
3.19 |
4.81 |
3.06 |
1.86 |
1.81 |
1.12 |
18.33 |
17.78 |
245631 |
245361 |
| 2 |
0.42 |
0.47 |
3.73 |
3.82 |
0.75 |
0.86 |
1.20 |
1.70 |
2.00 |
2.21 |
0.82 |
0.80 |
8.91 |
9.06 |
254631 |
254361 |
| 3 |
0.73 |
0.55 |
2.81 |
2.58 |
1.22 |
1.19 |
2.23 |
2.13 |
1.69 |
1.40 |
0.91 |
1.20 |
9.60 |
9.04 |
245361 |
245631 |
| 4 |
0.96 |
1.00 |
1.71 |
1.37 |
0.64 |
0.76 |
1.18 |
1.17 |
0.69 |
0.56 |
0.43 |
0.37 |
5.62 |
5.33 |
241536 |
241356 |
| 5 |
1.10 |
1.33 |
5.81 |
5.24 |
1.32 |
1.88 |
2.53 |
2.23 |
2.58 |
2.94 |
1.63 |
1.68 |
14.97 |
15.30 |
254631 |
254361 |
| 6 |
0.22 |
0.21 |
1.79 |
1.66 |
0.35 |
0.42 |
0.60 |
0.55 |
0.93 |
0.83 |
0.38 |
0.63 |
4.27 |
4.31 |
254631 |
256431 |
| 7 |
2.31 |
1.99 |
1.20 |
1.16 |
1.57 |
1.40 |
1.72 |
2.16 |
0.75 |
0.76 |
1.14 |
1.31 |
8.70 |
8.78 |
143265 |
416325 |
| 8 |
0.93 |
1.13 |
1.59 |
1.80 |
0.67 |
1.04 |
1.16 |
1.06 |
0.70 |
0.79 |
0.54 |
0.84 |
5.59 |
6.66 |
241536 |
214365 |
| 9 |
1.32 |
1.14 |
5.67 |
5.35 |
2.12 |
2.24 |
2.74 |
2.37 |
3.83 |
4.13 |
2.62 |
2.09 |
18.29 |
17.32 |
254631 |
254361 |
| 10 |
0.25 |
0.21 |
2.10 |
1.95 |
0.43 |
0.46 |
0.73 |
0.76 |
1.17 |
1.66 |
0.58 |
0.50 |
5.26 |
5.54 |
254631 |
254631 |
| 11 |
2.50 |
2.54 |
1.35 |
1.30 |
1.41 |
1.86 |
2.00 |
2.40 |
0.77 |
0.77 |
1.20 |
1.09 |
9.22 |
9.96 |
143265 |
143265 |
| 12 |
0.91 |
0.90 |
1.55 |
1.23 |
0.43 |
0.34 |
1.15 |
1.14 |
0.73 |
0.88 |
0.51 |
0.36 |
5.28 |
4.84 |
241563 |
241563 |
| 13 |
1.40 |
1.47 |
6.60 |
6.16 |
1.93 |
1.37 |
2.99 |
2.82 |
3.44 |
3.80 |
2.17 |
2.47 |
18.52 |
18.10 |
254631 |
254613 |
| 14 |
0.31 |
0.23 |
2.50 |
2.53 |
0.40 |
0.44 |
0.87 |
0.82 |
1.41 |
1.46 |
0.57 |
0.52 |
6.06 |
6.00 |
254631 |
254631 |
| 15 |
1.51 |
1.31 |
2.92 |
2.79 |
1.85 |
1.43 |
2.21 |
2.06 |
0.98 |
1.04 |
1.47 |
1.96 |
10.94 |
10.59 |
243165 |
246315 |
| 16 |
0.93 |
0.91 |
1.41 |
1.58 |
0.70 |
1.13 |
1.11 |
1.15 |
0.90 |
1.08 |
0.63 |
0.72 |
5.69 |
6.56 |
241536 |
243516 |
| 17 |
0.64 |
0.57 |
4.41 |
4.16 |
0.24 |
0.73 |
1.58 |
1.97 |
0.74 |
1.06 |
0.75 |
0.95 |
8.37 |
9.45 |
246513 |
245631 |
| 18 |
0.27 |
0.43 |
1.79 |
1.83 |
0.77 |
0.78 |
2.12 |
1.74 |
1.28 |
1.37 |
0.93 |
1.25 |
7.15 |
7.39 |
425631 |
245631 |
| 19 |
0.22 |
0.22 |
1.30 |
1.07 |
0.98 |
1.12 |
2.12 |
2.13 |
0.96 |
0.94 |
0.73 |
0.76 |
6.31 |
6.24 |
423561 |
432561 |
| 20 |
0.48 |
0.19 |
0.62 |
0.88 |
0.75 |
0.90 |
1.26 |
0.96 |
0.35 |
0.67 |
0.1 |
0.59 |
3.56 |
4.19 |
432516 |
432561 |
| 21 |
1.71 |
1.48 |
4.61 |
4.25 |
2.10 |
2.14 |
5.15 |
5.50 |
1.47 |
1.28 |
0.98 |
0.63 |
16.02 |
15.29 |
423516 |
423156 |
| 22 |
0.69 |
0.61 |
3.81 |
3.15 |
1.59 |
1.42 |
3.51 |
4.19 |
1.14 |
0.87 |
0.89 |
0.47 |
11.62 |
10.70 |
243561 |
423516 |
| 23 |
1.40 |
0.73 |
2.08 |
2.62 |
2.07 |
2.41 |
3.90 |
4.46 |
0.10 |
0.16 |
0.39 |
0.43 |
9.93 |
10.81 |
423165 |
423165 |
| 24 |
0.40 |
0.43 |
2.25 |
1.34 |
1.22 |
1.21 |
2.25 |
3.20 |
0.05 |
0.10 |
0.55 |
0.13 |
6.72 |
6.42 |
423615 |
423165 |
| Average |
0.96 |
0.88 |
2.98 |
2.79 |
1.10 |
1.22 |
2.04 |
2.23 |
1.32 |
1.36 |
0.95 |
0.95 |
9.36 |
9.40 |
|
|
| R2 |
0.90 |
0.97 |
0.78 |
0.89 |
0.89 |
0.75 |
0.98 |
|
|
|
|
|
|
|
|
|
Conclusion
The FTIR-PLS calibration model using 2nd derivative spectra and mean centering data processing, and a calibration set of 50 standard mixtures of the six kavalactones enabled qualitative and qualitative analyses of the content of the six kavalactones in kava samples. The results showed good analytical precision and accuracy with low errors (R2, 0.81–0.99; SEC, 0.72–1.73; SEP, 0.98–3.52). The FTIR-PLS predicted values of the six kavalactones in actual kava samples correlated very well with those by GC (R2, 0.75–0.98). The developed FTIR-PLS model can be used as an effective analytical tool to determine the content of six kavalactones in kava samples and thereby the chemotype. The selection of an appropriate spectral range between 1100 cm−1 and 1800 cm−1 can reduce detection errors due to inclusion of sufficient spectral information. The FTIR method is expected to complement traditional methods for the analysis of kava samples as well as other foods and beverages, which require special skills, and tedious sample pre-treatment.
Acknowledgements
We thank Dr C. S. Tang for his comments on this manuscript. This work was supported in part by grants from the State of Hawaii Department of Agriculture and Department of Health, USDA TSTAR (2005-34135-15989), and the University of Hawaii Research Council.
References
- Y. N. Singh, Kava: An overview, J. Ethnopharmacol., 1992, 37, 13–45 CrossRef CAS.
- H. Foo and J. Lemon, Drug Alcohol Review, 1997, 16, 147–155 Search PubMed.
- T. F. Munte, H. J. Heinze, M. Matke and J. Steitz, Neuropsychobiology, 1993, 27, 46–53 CrossRef CAS.
- H. R. W. Dharmaratne, N. P. Dhammika-Nanayakkaraa and I. A. Khan, Phytochemistry, 2002, 59, 429–433 CrossRef CAS.
- V. S. Parmar, S. C. Jain, K. S. Bisht, R. Jain, P. Taneja, A. Jha, O. D. Tyagi, A. K. Prasad, J. Wengel, C. E. Olsen and P. M. Boll, Phytochemistry, 1997, 46, 597–673 CrossRef CAS.
- Y. Shao, K. He, B. Zheng and Q. Y. Zheng, J. Chromatogr., A, 1998, 825, 1–8 CrossRef CAS.
- M. Ganzera and I. A. Khan, Chromatographia, 1999, 50, 649–653 CAS.
- V. Lebot, D. J. McKenna, E. Johnston, Q. Y. Zheng and D. McKern, Economic Botany, 1999, 53, 407–418.
- P. Lasme, F. Davrieux, D. Montet and V. Lebot, J. Agric. Food Chem., 2008, 56, 4976–4981 CrossRef CAS.
- L. Gautz, P. Kaufusi, M. C. Jackson, H. C. Bittenbender and C. S. Tang, J. Agric. Food Chem., 2006, 54, 6147–6152 CrossRef CAS.
- J. Denery, K. Dragull, C. S. Tang and Q. X. Li, Anal. Chim. Acta, 2004, 501, 175–181 CrossRef CAS.
- X. He, L. Lin and L. Lian, Planta Med., 1997, 63, 70–74 CrossRef CAS.
- H. Haberlein, G. Boonen and M. Beck, Planta Med., 1997, 63, 63–65 CrossRef CAS.
- M. Duffield, D. D. Jameson, R. O. Lidgard, P. H. Duffield and D. J. Bourne, J. Chromatogr., A, 1989, 475, 273–281 CrossRef CAS.
- R. N. Duve, Analyst, 1981, 106, 160–165 RSC.
- R. L. Young, J. W. Hylin, D. L. Plucknett, Y. Kawano and R. T. Nakayama, Phytochemistry, 1966, 5, 795–798 CrossRef CAS.
- T. F. Jiang, Y. H. Wang, Z. H. Lv and M. E. Yue, J. Pharm. Biomed. Anal., 2007, 43, 854–858 CrossRef CAS.
- M. Gaub, C. H. Roseler, G. Roos and K. A. Kovar, J. Pharm. Biomed. Anal., 2004, 36, 859–864 CrossRef CAS.
- R. Bilia, L. Scalise, M. C. Bergonzi and F. F. Vincieri, J. Chromatogr. B, 2004, 812, 203–214 CrossRef CAS.
- E. A. M. De-Jong and J. Kaper, Netherlands Milk and Dairy J., 1996, 50, 35–51 Search PubMed.
- I. F. Duarte, A. Barros, I. Delgadillo, C. Almeida and A. M. Gil, J. Agric. Food Chem., 2002, 50, 3104–3111 CrossRef CAS.
- N. Dupuy, M. Meurens, B. Sombret, P. Legrand and J. P. Huvenne, Appl. Spectrosc., 1992, 46, 860–863 CAS.
- M. Lin, M. Al-Holy, S.-S. Chang, Y. Huang, A. G. Cavinato, D.-H. Kang and B. A. Rasco, Int. J. Food Microbiol., 2005, 105, 369–376 CrossRef CAS.
- J. Moros, A. Inon, M. Khanmohammadi, S. Garrigues and M. de la Guardia, Anal. Bioanal. Chem., 2006, 385, 708–715 CrossRef CAS.
- J. Tewari and J. Irudayaraj, J. Agric. Food Chem., 2004, 52, 3237–3243 CrossRef CAS.
- H. Vardin, A. Tay, B. Ozen and L. Mauer, Food Chem., 2008, 108, 742–748 CrossRef CAS.
- B. Ozen, I. Weiss and L. J. Mauer, J. Agric. Food Chem., 2003, 51, 5871–5876 CrossRef CAS.
- Tay, R. K. Singh, S. S. Khrisnan and J. P. Gore, LWT-Food Sci. Technol., 2002, 35, 99–103 CrossRef.
- S. Côté, C. Kor, J. Cohen and K. Auclair, Biochem. Biophys. Res. Commun., 2004, 322, 147–152 CrossRef.
- H. K. Khurana, S. J. Jun, I. K. Cho and Q. X. Li, Appl. Eng. Agric., 2008, 24(5), 631–636 Search PubMed.
- S. Sivakesava and J. Irudayaraj, J. Food Sci., 2001, 66, 787–792 CrossRef CAS.
- Esteban-Díez, J. M. González-Sáiz and C. Pizarro, Anal. Chim. Acta, 2004, 514, 57–67 CAS.
- Esteban-Díez, J. M. González-Sáiz, C. Sáenz-González and C. Pizarro, Talanta, 2007, 71, 221–229 CrossRef.
- P. Geladi and B. R. Kowalski, Anal. Chim. Acta, 1986, 185, 1–17 CrossRef CAS.
-
J. H. Duckworth, in Applied Spectroscopy: A Compact Reference for Practitioners., ed. J. Workman and A. W. Springsteen, Academic Press, San Diego, 1998, vol. 74, pp. 93–163 Search PubMed.
-
Yoshinori, O. Sachiko, Y. Masashi, S. Yukie, S. Hiroto, and U. Masayuki, Eur. Pat., 1 222 925, 2005.
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.