DOI:
10.1039/C004432F
(Paper)
Anal. Methods, 2010,
2, 461-469
Extraction of metals from aqueous systems employing capillary-channeled polymer (C-CP) fibers modified with poly(acrylic acid) (PAA)
Received
17th March 2010
, Accepted 17th March 2010
First published on
15th April 2010
Abstract
Polyethylene terephthalate (PET, polyester) capillary-channeled polymer (C-CP) fibers were employed as the base structure for surface modification to provide a sorbent for solid phase extraction (SPE) of aqueous cations. Surface functionalization with polyacrylic acid (PAA) was achieved using a “grafting to” approach, yielding a stable sorbent that has the potential of being employed over a large range of pH values and a variety of sample matrices. PAA was chosen for surface modification because the repeating acrylic acid monomeric unit, which contains a carboxylic acid, has the ability to bind metal ions. In a micropipette-tip SPE format, these PAA-functionalized C-CP fibers showed the ability to efficiently bind several metals (Cu2+, Cu+, Ni2+, Fe3+, UO22+), while elution could be achieved with extremely low concentrations of hydrochloric acid (1 ml of 0.005%), resulting in overall recoveries of greater than 88%. A single tip was carried throughout the entire study as a control, and showed overall recoveries averaging 96% over the course of 19 extraction experiments and a 12-month period, demonstrating the chemical stability of the sorbent material, and suggesting the potential of the C-CP fiber SPE tips to be used multiple times.
Introduction
Solid phase extraction (SPE) is a proven method for sample pretreatment, the removal of matrix interferences and contaminants, and the pre-concentration of analytes prior to trace analysis.1,2 SPE originated in the 1950s with sorbent systems designed from charcoal packed into cartridges for extraction of organic contaminants from aqueous samples.3 The applications of SPE showed significant growth in the 1970s, in parallel with high performance liquid chromatography (HPLC), as the use of silica-based sorbents evolved.3,4 A variety of fields employ SPE, including biological, geological, and food analysis, and is of importance in the analysis of heavy metals in the fields of environmental remediation and trace contaminant detection. The ability to detect metals in aqueous environmental samples is of great concern as it has been shown that, while some metals at appropriate concentrations are beneficial, trace-level exposure to toxic metals (or high concentrations of otherwise beneficial metals) can cause health problems.4–6 The application of SPE to environmental samples requires the ability to immobilize trace amounts of elements across a variety of sample matrices. The complexity of sample matrices has changed dramatically over time with increases in industrialization and population, and therefore increases in pollution.7,8 The types of aqueous samples that require testing and monitoring are numerous, including natural, potable, and industrial waste waters. Even among these, there are large disparities in the matrix (e.g., salt and fresh waters) that require different processing approaches prior to instrumental analysis.
As the diversity of SPE applications grows, the demand for improved extraction media drives research and development of new approaches.9 Silica-based resins are the most commonly used and commercially available SPE phases.9,10 However, silica-based sorbents exhibit several disadvantages resulting in limitations when employed as a sorbent phase, such as a narrow pH window of 4–7 and residual surface silanol groups leading to undesired interactions. The porous nature of the silica sorbents is a two-edged sword; providing very high specific surface areas (a reflection of equilibrium loading capacities) but poor adsorption/desorption kinetics and recoveries which can lead to carry over and sample contamination.11 Researchers have extended the boundaries of what types of sorbents are considered typical (i.e. silica) to non-conventional materials as unique as human hair and bone powder.9 Recent research efforts have focused towards inorganic sol-gels,12–14 polyurethane foams,15 polymer sorbents4,6,16 and functionalized silica resins17,18 to provide high binding capacity and/or metal-specific sorbents. Relevant to the work described here, both polymer and silica-based fibers have also been used as SPE media for a variety of analyte species.19–23 Key driving forces here are the inherent low cost of fiber production, the ease of device packing, and the diversity of potential surface chemistries that can be exploited for effective adsorption.
The development of sorbents that possess fast kinetics, large binding capacities, high recovery, are reusable, can be adapted to any type format (micropipette tip, syringe, column), and uses dilute and/or small amounts of elution solvents is a continuing theme.6,9 We describe here the initial evaluation of polyacrylic acid (PAA) derivatized capillary-channeled polymer (C-CP) fiber solid phase extraction (SPE) tips for metal ion binding. The C-CP fiber format is under investigation in this laboratory for use as stationary phases in HPLC and SPE, primarily for proteomics applications.24–28 The fibers are melt-extruded from common textile polymers including polypropylene, polyester, and nylon-6 in such a manner that they have eight channels running along the entire fiber length.29 As suggested in the SEM micrograph in Fig. 1, the structure of the fibers results in interdigitation of the “fingers” within the column format, resulting in efficient use of space and a high packing density, while maintaining very high solvent transport efficiencies. In fact, single fibers (as well as fiber bundles) have the inherent ability to wick solutions due to the capillary action within the channels. Important for SPE applications, they exhibit ∼3 times the surface area of a circular fiber of the same nominal diameter,26 though the specific surface area (2–5 m2 g−1) is low by typical SPE standards, but on par with monolithic materials used in chromatography and SPE.1 On the other hand, the non-porous C-CP fibers have improved mass transfer kinetics when compared to porous materials, thus there is more accessible surface area in a dynamic situation. It is also worthy to note that C-CP fibers have demonstrated increased recovery/decreased carryover for proteins in both HPLC and SPE applications.24,29 The chemical nature of the base polymers allows operation across wide pH ranges (1–14),26,27 making them applicable as an extraction media for a wide range of sample matrices. Finally, as evidenced by the extremely wide portfolio of textile dye chemistries, polymer fibers present a plethora of potential means of surface modifications to affect high selectivity in SPE applications.
The ability to control surface interactions is of great importance and can be exploited in C-CP fibers by choosing the base polymer (polypropylene, polyethylene terepthalate (PET, polyester), nylon-6) to affect π–π, hydrophobic, and electrostatic interactions to varying degrees, allowing for their use in different modes of HPLC and SPE separations. For example, the application of nylon-6 as a base polymer allows for charge-based separations of proteins as the end groups provide a zwitterionic surface.30 As one means to enhance the charge density of the C-CP fiber surface, functionalization using a “grafting-to” approach that deposits a polyacrylic acid (PAA) layer on the surface of PET has been employed.31 In the approach, polymer chains with reactive functionalities react with complementary functional groups located on the surface of the substrate to form a layer of covalently anchored macromolecules. PAA was chosen for surface modification due to the presence of acrylic acid monomeric units in the macromolecule which results in a high density of accessible carboxylate groups (pKa = 4.3–4.532–34) on the surface after the grafting. Previous reports in the literature have demonstrated the use of PAA for metal chelation.35–40 The PAA modification of C-CP fibers provides a high density of cationic binding sites that can be tailored based on the molecular weight of the grafted polymer and the time over which grafting is allowed to occur. In addition to their high density of charged groups, PAA-coated C-CP fibers are well suited for trace metal extraction as a result of their stability/robustness over a wide range of pH values, nonporous structure, and low leachable impurities.
Presented here is the initial application of PAA-modified C-CP fibers as a sorbent material for metal ion extraction from aqueous media. Specifically, the PAA-modified fibers are employed in solid phase extraction micropipette tips. Detailed experiments involve determination of metal binding capacity and the ability of the PAA-functionalized C-CP fiber tips to bind a variety of metals both in single and multi-element solutions. In addition, experiments were designed to study the result of varying parameters such as the rate of loading (sample flow rate), exposure time, and the strength of the eluent on the adsorption/desorption and recovery of copper (Cu2+) from aqueous solutions. We suggest that PAA-functionalized C-CP fibers are a versatile, inexpensive, reusable sorbent for SPE that allows for recoveries greater than 88% for metal ions in aqueous media.
Experimental
Chemicals and reagents
Stock metal ion solutions, 3μg mL−1 unless otherwise specified, were prepared from water soluble salts (chloride or nitrate) obtained from Acros (Fair Lawn, NJ) using milliQ water (18.2 MΩ/cm) derived from a NANOpure Diamond Barnstead/Thermolyne Water System (Dubuque, IA). Uranium solutions, 3μg mL−1, were prepared from uranium oxide dissolved in trace metal grade nitric acid (VWR (West Chester, PA)) and diluted to the appropriate volume with milliQ water. Poly(glycidyl methacrylate), PGMA (synthesized as described elsewhere41) and polyacrylic acid from Sigma Aldrich (St. Louis, MO, USA) were used for functionalization of the sorbent phase. Standards used for calibration and hydrochloric acid used for desorption of metals from the fiber tips were obtained from VWR (West Chester, PA).
Instrumentation
Metal ion concentration determinations were performed using a Jobin-Yvon Horiba Ultima 2 (Longjumeau, France) inductively coupled plasma-optical emission spectrometer (ICP-OES) (Edison, NJ) and Analyst 5.2 software. The manufacturer default settings were used for ICP control and are presented in Table 1. The ICP is equipped with a single view radial plasma, a Meinhard concentric glass nebulizer, a cyclonic spray chamber, and a 1.0m Czerny–Turner monochromator containing 2400 grooves mm−1 holographic grating. Emission acquisition occurred with an integration time of 0.5 s, five wavelength points and five replicates per sample. Typical analytical response characteristics for the analytes determined in this study are presented in Table 2.
Table 1 ICP-OES operation conditions
| Parameters |
Condition |
| Power/W |
1000 |
| Ar gas flow rate/L min−1 |
12.0 |
| Nebulizer/L min−1 |
0.02 at 1.0 bar |
| Sheath gas flow rate/L min−1 |
0.20 |
| Peristaltic pump speed (rpm) |
20.0 |
| Replicates |
5 |
Table 2 Analytical response characteristics for target elements by ICP-OES
| Element |
Wavelength/nm |
Response Function |
R2 |
Detection Limits/ng mL−1 |
| Cu |
223.008 |
y = 28161x + 2475 |
0.9998 |
11 |
| Ni |
221.647 |
y = 27689x + 724.3 |
0.9999 |
3.6 |
| Fe |
259.940 |
y = 265924x + 5354 |
0.9999 |
0.30 |
| U |
385.958 |
y = 23996x − 2939 |
0.9968 |
18 |
Functionalization process
C-CP fibers are obtained from Clemson University School of Materials Science and Engineering in spool lengths greater than a 1000 m.42 Fibers are manually wound onto a rotary counter with a circular frame to produce a fiber bundle28 which is taken through the functionalization process involving a PGMA anchoring layer.31 Surface functionalization can be performed on C-CP fibers, regardless of the base polymer, using a “grafting-to” approach.31 In the present experiments, polyethylene terephthalate (PET, polyester) is used as the base polymer as it is readily extruded into the C-CP format, and present multiple opportunities for surface functionalization. Atmospheric pressure plasma treatment, using a Harrick Scientific Corporation (Model PDC-32G) apparatus, of the PET C-CP fibers, at a power of 7 watts for 10 min creates reactive carboxyl and hydroxyl functional groups on the surface. An anchoring layer of PGMA was then placed on the activated surface of the fibers using a dip coating method.31 The fibers were dried in air followed by annealing at 60 °C for 90 min to promote chemical tethering of the PGMA to the activated fiber surface.31 The excess PGMA was then removed by rinsing with acetone. The PAA was deposited by wetting the fibers with an ethanolic solution of PAA of the desired molecular weight. In these experiments, two different molecular weights of PAA were investigated; 5,000 Da and 100,000 Da. PAA was reacted with PGMA-coated C-CP fibers at various conditions; specifically three combinations were explored in detail. A grafting time and temperature combination of 80 °C for 120 min provides a high density of functional groups while a combination such as 60 °C for 10 min provides a lower density of functional groups. After the desired grafting time was reached, the excess PAA was removed by rinsing with water. The combination of grafting density and molecular weights of PAA employed, yielded three combinations of functionalized C-CP fibers; high molecular weight-high grafting density (HMWHD), high molecular weight-low grafting density (HMWLD) and low molecular weight-high grafting density (LMWHD). The amount of the grafted to PET surface polymer was determined using ellipsometry for model PET films (deposited on silicon wafer) as described elsewhere.31 The ultimate metal binding capacity of the fibers is determined by the number of available binding sites, which is controlled by the functionalization conditions. As the molecular weight of the PAA increases, so to does the thickness of the layer and, therefore, more functional carboxylic groups are placed on the surface. In addition, as the grafting time is increased the grafting density of the polymer increases as well, increasing the amount of polymer placed on the fibers. Fig. 2 pictorially depicts the HMWHD, HMWLD, and LMWHD functionalized fibers and how metal binding may be affected by the structure of the PAA layer.
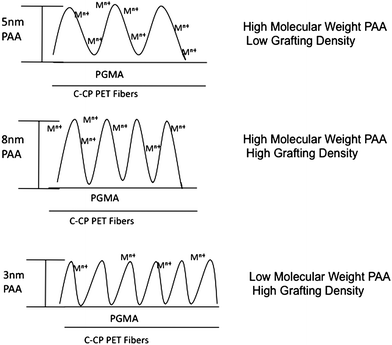 |
| | Fig. 2 Diagrammatic representation of the assumed binding site distributions for the three PAA coating conditions. | |
 |
| | Fig. 3 Photograph of C-CP fiber packed SPE tip attached to micropipette tip. | |
For the sake of consistency, all experimental data presented here was acquired for centrifugation performed using the same protocol. All of the test, rinse, and elution solutions were loaded in 1ml aliquots because the fiber packed tip is attached to a 1ml commercial pipette tip, yielding collected volumes sufficient to be analyzed on the ICP-OES. More specifically, 1 ml of sample solution was loaded into the tips and centrifuged for 5 min; this process was repeated 3 times for a total volume (Vt) of 3 ml. The tips were then rinsed and centrifuged with three, 1 ml aliquots of milliQ water, to remove non-specifically bound analyte. Finally, three 1 mL aliquots of the hydrochloric acid elution solutions were passed through the SPE tips for desorption of the immobilized metals from the surface, Vt = 3 ml. The entire analytical procedure described here, from loading of the sample to elution for ICP-OES analysis, takes 45 min to complete, though performed in parallel batches. RCFs ranging from 575–1900 were examined using a Clinical 50 Centrifuge (VWR, West Chester, PA), it was determined that the adsorption/desorption efficiencies remained relatively constant over this range as the percentage recoveries did not vary significantly. A nominal centrifugation rate of 1200 RCF, was quite sufficient to obtain quantitative recoveries, with the solution transient times under these conditions of ∼1 min, and so the total process could be completed in less than 10 min on sample batches.
A key variable when evaluating the PAA-functionalized C-CP fibers as an SPE sorbent is to determine the lifetime of the tips. Due to the efficiency of the acid elution process, there is no requirement for regeneration of the PAA-fiber surfaces prior to reuse of the tips, therefore the SPE tips are ready for loading of the next sample immediately following the acid elution step. Even so, the robustness of the polymer coating itself must be evaluated. A single control SPE tip was generated from a HMWHD functionalization of PAA, which was run under the exact same conditions, 3ml of 3 μg ml−1 Cu2+ loaded and elution with 5% HCl, in parallel with each experimental variation performed on the remaining experimental tips. The control tip's recovery was evaluated after each experiment to ensure that the data collected during each experimental set was valid, while also determining the stability of the PAA-functionalized C-CP fiber tips.
Results and discussion
Qualitative aspects
The PAA-functionalized surface provides anionic binding sites that should be capable of binding cationic metal species. Literature reports confirm that copper binds strongly to both linear and cross-linked polyacrylic acid polymer chains.36,38,43 Qualitative experiments were performed by soaking functionalized fibers in concentrated aqueous solutions of copper. When exposed to an aqueous Cu2+ solution, the fiber surfaces visibly showed that copper was bound to the surface. Fig. 4 is an optical photograph of PAA- functionalized C-CP fiber packed SPE tips that have bound Cu2+, which is visible by the blue-green surface coloration. To investigate whether the copper was bound electrostatically to the fiber surface, the fibers were subsequently rinsed with DI-H2O with no visual evidence in the wash solution that copper was released, nor was there a decrease in the intensity of the fiber color; suggesting that the Cu2+ was indeed bound to the surface functional groups. The fibers were subsequently treated with hydrochloric acid, resulting in removal of copper from the PAA- functionalized C-CP fiber surface apparent by the resulting blue green solution and white fiber surface.
 |
| | Fig. 4 Photograph of PAA-functionalized C-CP fiber tips with Cu2+ bound to the surface. | |
Each of the three variations of PAA functionalization (i.e. HMWHD, HMWLD, LMWHD) was evaluated to determine extent of the polymer surface coverage. Ellipsometry is a common approach used to characterize polymer layer thicknesses of chemically similar, dielectric materials.44 Because of the roughness of the C-CP fibers due to the presence of the channel structure, thickness measurements were performed instead on silica wafers covered with PET film and functionalized using the same “grafting-to” approach used on the C-CP fibers.31 Based on the determined layer thickness and the theoretical number of chains per unit length, the number of expected carboxylate groups per unit surface area can be calculated. From that number and the computed total fiber surface area of fiber in the tips, the binding capacity of Cu2+ can be projected based on an assumed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 Cu2+:carboxylate binding stoichiometry. A comparison of the anticipated binding capacity (mass) of Cu2+ based on calculations from the ellipsometry measurements and those determined experimentally through Cu2+ binding to three corresponding PAA-C-CP fiber tips (as determined by ICP-OES detection) is presented in Table 3.
:2 Cu2+:carboxylate binding stoichiometry. A comparison of the anticipated binding capacity (mass) of Cu2+ based on calculations from the ellipsometry measurements and those determined experimentally through Cu2+ binding to three corresponding PAA-C-CP fiber tips (as determined by ICP-OES detection) is presented in Table 3.
Table 3 Evaluation of binding sites on PAA C-CP SPE tips reported as amount of bound Cu2+
| Method |
HMWHD |
HMWLD |
LMWHD |
| Thickness/nm |
Mass/μg |
Thickness/nm |
Mass/μg |
Thickness/nm |
Mass/μg |
|
Values computed based on thickness of functional layer.
Values obtained by SPE using ICP-OES detection.
|
| Ellipsometery |
8 |
7.7a |
5 |
4.7a |
3 |
3.0a |
| SPE |
— |
6.9b |
— |
5.5b |
— |
3.8b |
As can be seen, the values are in quite good agreement. Discrepancies between theory and SPE binding measurements can be explained by taking into consideration that SPE looks at carboxyl groups actually accessible to Cu2+ for binding and the actual coordination of Cu2+ to PAA on the fiber surface. The ellipsometry-based calculations assume that all carboxylate groups are available for binding, that the calculated number is the same as the actual number of groups present on the surface, and that a 1:2 Cu2+:carboxylate coordination occurs. However, significant fraction of acrylic acid groups (responsible for PAA anchoring to the surface via reaction with epoxy groups in the PGMA chain) is not available for the attachment of Cu2+. It is also known that Cu2+ coordinates with carboxylate groups; as bi-dentate or higher-order complexes.37 However, the extent to which coordination occurs at a surface depends on conditions, such as the distance between surface ligands and the pH of the solution.37,38 The coordination of Cu2+ to these specific PAA- functionalized C-CP fiber surfaces has not been studied, but PAA complexation of Cu2+ in solution has been studied and was determined that at a minimum of two carboxylates are involved per metal ion (i.e. bi-dentate).37 Since the coordination assumptions made to perform the ellipsometry calculations (i.e., 1:2 Cu2+:carboxylate) and the number of available carboxylates may not be completely valid, it makes sense that the computed loading masses are biased-high in comparison to the actual binding seen in the SPE numbers.
Quantification
Experiments were designed to focus on optimizing the binding/elution conditions, determination of binding capacity, metal-specific binding vs. competitive binding, and process recovery. The metal ion solutions were loaded, tips rinsed with DI-H2O, and analyte eluted with hydrochloric acid in three 1 mL fractions as previously described. The recovered loading solution (i.e., the test solution passed through the tip) was analyzed to determine the total amount of metal ions bound to the surface by calculating the difference between the stock solution and the flow through from the loading solution. The rinse solution was analyzed for non-specifically bound metal ions and the acid eluate solution analyzed for specifically bound metal ions.
Control tip
One of the most outstanding attributes of the PAA-functionalized C-CP fiber SPE tips is that the cost associated with the fiber in a single tip is only ∼$0.03 US. In addition, the capillary packing and micropipette tip fitting is quite simple in comparison to that of bead-form sorbents. Even so, it would be beneficial to be able to use single tips through multiple metal ion extractions and to know the extent to which there is carryover or polymer degradation following acid elution. In order to test these potentialities, and to ensure that the results obtained throughout the course of the experiments were valid and not related to exhaustion of the tip lifetime, a single control tip was analyzed repeatedly under identical conditions throughout the course of this study. The control tip was made from HMWHD PAA-functionalized C-CP fibers and was analyzed each time an experimental variable was altered and studied (i.e., a new experimental set, as denoted by the name). A 3.0 μg mL−1 Cu2+ solution, applied in three 1 mL aliquots (a total of 9 μg of Cu2+) was used as the analyte in the control tip. (The sole exceptions to this method were in the case of the competitive binding experiments where 0.9 μg of Cu2+ was applied.) Based on preliminary studies, this amount of Cu was enough to overload the PAA SPE micropipette tips. Elution was accomplished using three 1 ml aliquots of a 5% HCl elution solvent, instead of the lower HCl concentrations used for elution in all other experiments. The use of 5% HCl represents an extreme case in terms of testing the chemical robustness of the adsorbed PAA layer as well as the physical structure of the fiber-packed tip. Observation of decreased recoveries here would have allowed the tips to be replaced before decreased efficiency actually affected those other experiments. Table 4 summarizes the experimental sets for which the control tip was run in conjunction, the amount of metal bound to the fibers, the efficiency of that process (% Cu2+ adsorbed), and the efficiency of the desorption of the captured metal (% Cu2+desorbed). As can be seen the % Cu2+adsorbed in each experiment is very consistent, averaging 87.3% with a variation of 6.4%RSD across the 19 exposures. This number is less than 100% because the tips were intentionally, slightly overloaded. The control SPE tip averaged a 97% overall recovery of the adsorbed Cu2+ over 19 applications and showed no signs of either chemical or physical degradation over a 12 month period. These results clearly illustrate the stability of the PAA-functionalized C-CP fiber SPE phases, even under the fairly challenging 5% HCl elution conditions and indeed drying in between experiments. As such, there is a high degree of confidence of the relative validity of the results obtained across the range of the various test conditions.
Table 4 Control HMWHD on PAA-Functionalized C-CP SPE tips as determined by ICP-OES detection
| Experimental Seta |
Mass Adsorbed/μg |
% Cu2+ Adsorbed |
% Cu2+ Desorbed |
|
Experimental parameter that was varied to correspond to control tip data.
|
| 5.0% HCl |
6.9 |
83 |
100 |
| 0.5% HCl |
8.3 |
85 |
94 |
| 0.1% HCl |
7.1 |
85 |
100 |
| 0.05% HCl |
8.1 |
80 |
93 |
| 0.1% HCl |
7.9 |
80 |
97 |
| 0.005% HCl |
8.0 |
85 |
95 |
| 0.001% HCl |
7.7 |
85 |
96 |
| Ni2+ Run 1 |
8.7 |
89 |
100 |
| Ni2+ Run 2 |
8.5 |
90 |
97 |
| Cu+ Run 1 |
7.4 |
90 |
98 |
| Cu+ Run 2 |
7.3 |
90 |
100 |
| UO22+ Run 1 |
7.1 |
81 |
94 |
| UO22+ Run 2 |
8.9 |
89 |
100 |
| Fe3+ Run 1 |
7.8 |
100 |
100 |
| % Recovery Run 1 |
7.8 |
88 |
97 |
| % Recovery Run 2 |
8.1 |
80 |
88 |
| Competitive Binding Run 1 |
0.84 |
93 |
93 |
| Competitive Binding Run 2 |
0.90 |
97 |
97 |
| % Recovery Run 3 |
7.8 |
88 |
98 |
Cu2+ elution characteristics
Elution of metal from the PAA surface takes place by employing hydrochloric acid, so that the pH decreases below the pKa of the PAA resulting in protonation of the carboxylate functional groups on the fiber surface, displacing the metal ions into solution. In order to make an SPE application more environmentally friendly and consumer affordable, low concentrations/low volumes of elution solvent are preferable. Three 1 ml aliquots of the 3 μg ml−1 solution of CuCl2 were run through the tips, exposing a total of 9 μg Cu2+ to the fiber surface, which had been previously determined to be in excess of the binding capacity of the amount of fiber in the present tips. The tips were exposed to the excess to ensure that the full binding capacity of the tips was achieved. The respective solutions were passed through the tips using centrifugal force of 1215 RCFs. The eluting acid concentrations were varied between 0.001% and 5% (in 3, 1 ml aliquots) in order to evaluate the minimum solvent strength necessary for elution of Cu2+ ions from the three densities of PAA-functionalized C-CP SPE tips. The experimental results presented in Table 5 show that there is no difference within experimental error for elutions performed in the range 0.005%-to-5% HCl for the HMWHD and HMWLD polymers. In the case of LMWHD coating, the amount of copper desorbed from the surface varies with the elution strength, but follows no discernible trends. Statistical analysis (student t-test), at a 95% confidence interval, of the LMWHD recoveries showed no significant difference among the solvent elution strengths (i.e. HCl concentrations). It is also clear that the HMWHD PAA fibers, covered with the thickest PAA layer, showed the greatest amount of variability within experimental sets. We suppose that the observed phenomenon can be connected to limited solubility (swellability) of the grafted PAA chains in the water with acidic pH. Therefore, for the relatively thick grafted layer diffusion limited, non-equilibrium extraction may occur. This phenomenon will require further investigation. When elution is attempted with 0.001% HCl, approximately 60% recovery occurs in HMWHD and HMWLD, with a second elution of 5% HCl allowing complete recovery. Statistically there is no dissimilarity in the recovery with varying HCl strength until the 0.001% HCl was used with the exception of a single outlier in both the HMWHD and HMWLD cases (at 0.5% and 0.05%, respectively). The 5% upper limit of exposure was chosen because the stability of PAA on the C-CP fibers had not been evaluated above this point. In addition, the amount of copper exposed to the surface can be accounted for with recoveries >90% when 5% HCl was used for elution. Therefore, it was unnecessary to explore higher concentrations of HCl as an increase in cost and potential degradation of the functionalized surface layer are all negatives that will not be offset by a substantial increase in recovery. However, this study only looked at the strength of elution solvent necessary to elute Cu2+ from the surface; other metals may need higher concentrations, stronger acids, or a chelator to affect efficient removal.
Table 5 Desorption of Cu2+ from the surface of PAA-Functionalized C-CP SPE tips as determined by ICP-OES detection
| % HCl |
HMWHD/μg |
HMWLD/μg |
LMWHD/μg |
| 5 |
6.9 ± 0.9 |
5.5 ± 0.2 |
3.8 ± 0.2 |
| 0.5 |
8.1 ± 2.5 |
5.2 ± 0.3 |
5.2 ± 0.3 |
| 0.1 |
6.9 ± 1.7 |
5.3 ± 0.2 |
5.3 ± 0.2 |
| 0.05 |
6.2 ± 1.3 |
3.9 ± 1.2 |
3.9 ± 0.2 |
| 0.005 |
5.8 ± 2.3 |
5.4 ± 0.3 |
5.4 ± 0.3 |
| 0.001 |
3.9 ± 1.1 |
3.8 ± 0.4 |
3.8 ± 0.4 |
Metal species' binding characteristics
In order to begin to evaluate the performance of the PAA-functionalized C-CP fiber tips for metal ion extractions from complex matrices, several factors need to be explored including the ability to bind a variety of metal ions, the extent of competitive binding, and the total recovery of metals. Several transition metals were evaluated, in individual solutions, for their binding capacities to the PAA-functionalized fiber tips; Cu2+, Cu+, Ni2+, Fe3+, and the lanthanide oxide complex, UO22+. Table 6 lists the metal loading/recovery for the three PAA surface layers for the metals initially applied as three 1 ml aliquots of 3 μg mL−1 concentration solutions, resulting in a total of 9 micrograms exposed. There is an initial trend that is obvious regardless of the metal bound to the surface; the HMW PAA tips have the highest binding capacities. In fact the higher molecular weight grafted layers are thicker, therefore more functional groups are available for binding. The relative binding capacities for the individual metal ions varied, though the relative amounts for each metals were consistent between the PAA C-CP fiber surface types. The concept of hard soft acid base theory (HSAB) can help explain some of the binding trends among the metal ions. Carboxylate ions (the ligand species on the PAA surface) are considered hard bases. On the other hand, PAA is a weak polyelectrolyte, that as the molecular weight of the polymer increases becomes a weaker acid, i.e. borderline base.37,40 The metals investigated are a combination of soft (Cu1+), borderline (Ni2+, Cu2+), and hard (Fe3+,UO22+) character. The best binding capacity comes with Cu2+, a borderline metal, in all three degrees of functionalization. The higher level of complexation of Cu2+ with the –COO− surface ligands of PAA is also in line with the Irving Williams series relationship.45 Overall, the differences observed in the binding capacity of the remaining metals are believed to be related to some combination of the extent of metal complexation (i.e. mono-, bi-dentate, etc. with PAA ligands), coordination geometry, and hydration; all factors that need to be explored in further detail. The differences observed here are most surely in the adsorption step as opposed to differences in the elution characteristics as the elution recoveries and mass balance values are quite uniform.
Table 6 Metal Concentration on PAA-Functionalized C-CP SPE tips as determined by ICP-OES detection
|
|
HMWHD/μg |
HMWLD/μg |
LMWHD/μg |
| Cu2+ |
6.9 ± 0.9 |
5.5 ± 0.2 |
3.8 ± 0.2 |
| Cu1+ |
5.5 ± 1.1 |
4.8 ± 0.8 |
4.2 ± 0.2 |
| Ni2+ |
2.1 ± 0.6 |
1.7 ± 0.1 |
0.76 ± 0.02 |
| Fe3+ |
2.9 ± 1.1 |
2.4 ± 0.3 |
2.5 ± 0.2 |
| UO22+ |
2.8 ± 0.8 |
1.9 ± 0.4 |
1.2 ± 0.4 |
Table 7 shows a comparison of the PAA functionalized C-CP fiber tips to other silica and polymer based metal sorbents. The capacities reported in Table 7 for all sorbent phases are based on Cu2+ binding. The binding capacity of the HMWHD PAA fibers is comparable to the other fiber sorbent phase, polyacrylonitrile based, presented. Most of the sorbent materials presented in Table 7 are small and porous particles whereas the PAA sorbent exhibits lower binding capacity due to the nonporous nature of the fibers (resulting in much lower (single m2 g−1) specific surface area). Comparisons of total binding sites on each surface (which are probably the greatest determining factor) are difficult to make based on the available literature. On the other hand, the non-porous nature of the fibers results in increased mass transfer efficiencies during the adsorption desorption steps as well as puts much lower backpressure constraints in terms of fluidics than in the case of pack-bed media. These latter two characteristics are of high relevance in high throughput analyses.
Table 7 Comparison of PAA-Functionalized C-CP fibers with other sorbent phases in terms of Cu2+ binding
|
|
Capacity/μmol g−1 |
Sorbent Base Material |
Particle Size/μm |
Sorbent Format |
Analyte Volume/mL |
Adsorption Time/min |
Total Extraction Time/min |
| 3-methyl-1-phenyl-4-stearoyl-5-pyrazolone |
43 |
Silica Gel |
149–420 (70 Å) |
Column |
1000 |
200 |
208 |
| Salicylaldoxime |
80 |
Silica Gel |
60 (120 Å) |
Column |
25 |
3 |
28 |
| Resacetophenone |
186 |
Silica Gel |
125–250 |
Column |
50 |
15–50 |
65–225 |
|
LMWHD PAA
|
17
|
PET Polymer Fibers
|
— |
Micropipette Tip
|
3
|
3–15 |
9–45 |
|
HMWLD PAA
|
25
|
PET Polymer Fibers
|
— |
Micropipette Tip
|
3
|
3–15 |
9–45 |
|
HMWHD PAA
|
31
|
PET Polymer Fibers
|
— |
Micropipette Tip
|
3
|
3–15 |
9–45 |
| Poly(acrlyaminophosphonic-dithiocarbamate) |
32 |
Polyacrylonitrile Fiber |
— |
Column |
1000 |
200 |
210 |
|
o-Aminophenol |
53 |
Amberlite XAD-2 (polystyrene copolymer) |
250–841 (90 Å) |
Column |
1000 |
50 |
510 |
| Chromotropic Acid |
134 |
Amberlite XAD-2 (polystyrene copolymer) |
250–841 (90 Å) |
Column |
100 |
33–50 |
70–105 |
| Iminodiacetic Acid 3M Empore |
1900 |
Poly (styrenedivinylbenzene) |
8 |
Disk |
100 |
1 |
24 |
Metal species selectivity
The single solution binding experiments showed the tips had an appreciably higher capacity for Cu2+ ions. In order to explore the potential selectivity/bias of the PAA-functionalized C-CP fiber tips, experiments were performed using Cu2+ and Ni2+ solutions. The loading solution was prepared to contain equal amounts of copper and nickel, 0.9 μg each. This total mass is appreciably below the maximum capacity of the tips to prevent overloading. Table 8 shows both the flow through (unbound metal) and the recovered amounts for Cu2+ and Ni2+ for the mixed-metal solutions. It is apparent that when the SPE tips are presented with a dual element solution the tips exhibit a preference for Cu2+, as all 0.9 μg applied was bound/eluted from each of the fiber tip surfaces. On the other hand, there is appreciable flow-through of the Ni2+ species, for each of the tips. That said, those amounts of Ni2+ adsorbed are recovered in the elution step. Clearly, one must consider both kinetic and thermodynamic effects here, and so static adsorption studies are in order to better understand adsorption biases.
Table 8 Metal Concentration on PAA-Functionalized C-CP SPE tips as determined by ICP-OES detection
|
|
Flow Through/μg |
Desorption/μg |
| Cu2+ |
Ni2+ |
Cu2+ |
Ni2+ |
| HMWHD |
0.00 ± 0.00 |
0.37 ± 0.14 |
0.88 ± 0.04 |
0.43 ± 0.14 |
| HMWLD |
0.02 ± 0.04 |
0.57 ± 0.06 |
0.89 ± 0.09 |
0.24 ± 0.06 |
| LMWHD |
0.01 ± 0.01 |
0.71 ± 0.03 |
0.96 ± 0.11 |
0.13 ± 0.02 |
Recovery
In order for the PAA-functionalized C-CP fibers to be useful in applications such as trace contaminant detection, it is important that they can process large volumes of low concentration metals, concentrating them through elution into smaller volumes. In order to evaluate this ability, 10 ml of a dilute solution of Cu2+ was run through the tips, totaling 2.2 μg and 4.4 μg for Trial 1 and 2 respectively. The tips were then washed with water, followed by elution with a total of 3 ml of 1% HCl, with analysis performed on the eluate by ICP-OES. This experiment was repeated twice (trial 1 and 2) and results are presented in Table 9. This data shows that with the currently utilized conditions all tips are capable of producing recoveries of at least 88%.
Table 9 % Recovery of Cu2+ on PAA-Functionalized C-CP SPE tips as determined by ICP-OES detection
|
|
Flow Through/μg |
Amount Desorbed/μg |
% Recovery |
| Trial 1 (2.2 μg loaded) |
| HMWHD |
00.0 ± 0.00 |
2.8 ± 0.0 |
104 |
| HMWLD |
00.0 ± 0.00 |
1.8 ± 0.2 |
92 |
| LWMHD |
00.0 ± 0.00 |
2.0 ± 0.2 |
100 |
| Trial 2 (4.5 μg loaded) |
| HMWHD |
0.05 ± 0.05 |
4.6 ± 0.4 |
102 |
| HMWLD |
0.21 ± 0.11 |
3.8 ± 0.2 |
88 |
| LMWHD |
0.26 ± 0.06 |
3.8 ± 0.3 |
90 |
Conclusions
The functionalization of polyester C-CP fibers with polyacrylic acid (PAA) results in a high density, anionic surface capable of metal binding. It is evident from the results presented here that binding and elution of several metals from the SPE tips surface can occur with low concentrations of HCl, resulting in an environmentally safe and cheap method for metal detection. The non-porous structure of the C-CP fibers shows its advantages in the fast binding kinetics and high recoveries of analyte from the tips. The C-CP fibers were functionalized with a high molecular weight (HMW) and a low molecular weight (LMW) PAA, after evaluation of the tips for copper binding it is apparent that the HMW coated tips have a higher density of functional groups. When exposed to dual-element solutions the PAA functionalized C-CP fiber tips exhibit a binding preference for Cu2+. PAA functionalized C-CP fiber tips are low cost (<$0.03/tip) and reusable as was demonstrated in the ability to use the fiber tips for 19 experiments over a 1 year period while still maintaining 96% recovery on the control tip. Future experiments will look at the role of adsorption conditions, including static studies, to better understand the binding of individual metals and any preferential binding that occurs. Expansion of the desorption studies to include other eluents (i.e. complexation agents, EDTA) will also be undertaken. On a more fundamental level, experiments to investigation the surface interactions of the metals with the functionalized surface, i.e. PAA-Mn+ complexation stoichiometry, will be performed. Finally, given the high fluidic transport efficiencies of the functionalized C-CP fibers, use of flow-through loops and cartridges seems to be an area of great potential.
Acknowledgements
Financial support from the Clemson University Center for Advanced Engineering Fibers and Films (CAEFF), a National Science Foundation Engineering Research Center operating under grant No. EEC-9731680 is gratefully appreciated.
References
-
S. Xie, T. Jiang, F. Švec and R. W. Allington, in Journal of Chromatography Library Monolithic Materials Preparation, Properties, and Applications, ed. F. Švec, T. D. Tennikova and Z. Deyl, Elsevier Science B.V., Amsterdam, The Netherlands. 2003, vol. 67, pp. 687–697 Search PubMed.
- R. E. Majors, LC-GC North America, 2008, 26, 1074–1090 CAS.
- I. D. W. Colin and F. Poole, J. Chromatogr., A, 2000, 885, 1 CrossRef.
- I. Dakova, I. Karadjova, I. Ivanov, V. Georgieva, B. Evtimova and G. Georgiev, Anal. Chim. Acta, 2007, 584, 196–203 CrossRef CAS.
- B. Pan, Q. Zhang, W. Du, W. Zhang, B. Pan, Q. Zhang, Z. Xu and Q. Zhanga, Water Res., 2007, 41, 3103–3111 CrossRef CAS.
- B. Rezaei, E. Sadeghi and S. Meghdadi, J. Hazard. Mater., 2009, 168, 787–792 CrossRef CAS.
- F. L. Melquiades, P. S. Parreira, M. J. Yabe, M. Z. Corazza, R. Funfas and C. R. Appoloni, Talanta, 2007, 73, 121–126 CrossRef CAS.
- I. Liska, J. Chromatogr., A, 2000, 885, 3–16 CrossRef CAS.
- A. R. Türker, Clean, 2007, 35, 548–557 Search PubMed.
- S. Risticevic, V. H. Niri, D. Vuckovic and J. Pawliszyn, Anal. Bioanal. Chem., 2009, 393, 781–795 CrossRef CAS.
-
U. D. Neue, HPLC Columns Theory, Technology, and Practice, Wiley-VCH, New York. 1997 Search PubMed.
- P. Misaelides, G. Gallios, S. Sarri, D. Zamboulis, E. Pavlidou, N. Kantiranis, I. Anousis, I. Zhuravlev and V. V. Strelko, Sep. Sci. Technol., 2006, 41, 97–110 CrossRef CAS.
- B. Saad, C. C. Chong, A. S. M. Ali, M. F. Bari, I. A. Rahman, N. Mohamad and M. I. Saleh, Anal. Chim. Acta, 2006, 555, 146–156 CrossRef CAS.
- I. Z. Zhuravlev, V. A. Kanibolotsky, V. V. Strelko and G. P. Gallios, Sep. Sci. Technol., 2005, 39, 287–300 CrossRef.
- V. A. Lemos, M. S. Santos, E. S. Santos, M. J. S. Santos, W. N. L. d. Santos, A. S. Souza, D. S. d. Jesus, C. F. d. Virgens, M. S. Carvalho, N. Oleszczuk, M. G. R. Vale, B. Welz and S. L. C. Ferreira, Spectrochim. Acta, Part B, 2007, 62, 4–12 CrossRef.
- K. Oshita, A. Sabarudin, T. Takayanagi, M. Oshima and S. Motomizu, Talanta, 2009, 79, 1055–1060 CrossRef CAS.
- X. Zhu, X. Chang, Y. Cui, X. Zou, D. Yang and Z. Hu, Microchem. J., 2007, 86, 189–194 CrossRef CAS.
- M. H. Mashhadizadeh, M. Pesteh, M. Talakesh, I. Sheikhshoaie, M. M. Ardakani and M. A. Karimi, Spectrochim. Acta, Part B, 2008, 63, 885–888 CrossRef.
- L. Q. Chen, G. Yang and J. Zhang, React. Funct. Polym., 1996, 29, 139–144 CrossRef CAS.
- A. M. C. Freitas, C. Parreira and L. Vilas-Boas, Chromatographia, 2001, 54, 647–652.
- K. Jinno, Y. Saito and M. Imaizumi, Bunseki Kagaku, 2001, 50, 775–783 CrossRef CAS.
- Y. Liu, M. L. Lee, K. J. Hageman, Y. Yang and S. B. Hawthorne, Anal. Chem., 1997, 69, 5001–5005 CrossRef CAS.
- M. Radetic, D. Jocic, P. Jovancic, L. Rajakovic, H. Thomas and Z. L. Petrovic, J. Appl. Polym. Sci., 2003, 90, 379–386 CrossRef CAS.
- R. K. Marcus, W. C. Davis, B. C. Knippel, L. LaMotte, T. A. Hill, D. Perahia and J. D. Jenkins, J. Chromatogr., A, 2003, 986, 17–31 CrossRef CAS.
- D. K. Nelson and R. K. Marcus, J. Chromatogr. Sci., 2003, 41, 475–479.
- D. M. Nelson and R. K. Marcus, Anal. Chem., 2006, 78, 8462–8471 CrossRef CAS.
- D. M. Nelson and R. K. Marcus, Protein Pept. Lett., 2006, 13, 95–99 CrossRef CAS.
- D. S. Fornea, Y. Wu and R. K. Marcus, Anal. Chem., 2006, 78, 5617–5621 CrossRef CAS.
- W. A. Haile and B. M. Phillips, Tappi J., 1995, 78, 139–142 Search PubMed.
-
C. Straut, M. Permaul and R. K. Marcus, Pittsburgh Conference, Chicago, IL, Year Search PubMed.
- O. Burtovyy, V. Klep, H.-C. Chen, R.-K. Hu, C.-C. Lin and I. Luzinov, J. Macromol. Sci., Part B: Phys., 2007, 46, 137–154 Search PubMed.
- S. Chibowski and M. Paszkiewicz, Physicochem. Prob. Min. Process., 2006, 40, 175–184 Search PubMed.
- J. E. Gebhardt and D. W. Fuerstenau, Colloids Surf., 1983, 7, 221–231 CrossRef CAS.
- D. G. Leaist, J. Solution Chem., 1989, 18, 421–435 CAS.
- H. P. Gregor, L. B. Luttinger and E. M. Loebl, J. Phys. Chem., 1955, 59, 990–991 CAS.
- H. P. Gregor, L. B. Luttinger and E. M. Loebl, J. Phys. Chem., 1955, 59, 34–39 CrossRef CAS.
- R.-S. Juang and J.-F. Liang, J. Membr. Sci., 1993, 82, 163–174 CrossRef CAS.
- A. M. Kotliar and H. Morawetz, J. Am. Chem. Soc., 1955, 77, 3692–3695 CrossRef CAS.
- B. L. Rivas and I. Moreno-Villoslada, Polym. Bull., 1997, 39, 653–660 CrossRef CAS.
- B. L. Rivas, L. N. Schiappacasse, U. E. Pereira and I. Moreno-Villoslada, Polymer, 2004, 45, 1771–1775 CrossRef CAS.
- B. Zdyrko, K. S. Iyer and I. Luzinov, Polymer, 2006, 47, 272–279 CrossRef CAS.
- D. M. Nelson, R. D. Stanelle, P. Brown and R. K. Marcus, Am. Lab., 2005, 37, 28 CAS.
- H. P. Gregor, L. B. Luttinger and E. M. Loebl, J. Phys.l Chem., 1955, 59, 366–368 Search PubMed.
-
H. Fujiwara, Spectrosocpic Elipsometry: Principles and Applications, John Wiley & Sons, New York. 2007 Search PubMed.
-
J. E. Huheey, E. A. Keiter and R. L. Keiter, Inorganic Chemistry: Principles of Structure and Reactivity, HarperCollins College Publisers, New York. 1993 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here. 