A simple strategy for preparation of spherical silica-supported porous chitosan matrix based on sol–gel reaction and simple treatment with ammonia solution
Xingyong
Xu
a,
Pingjun
Dong
b,
Yan
Feng
b,
Feng
Li
*b and
Hongjun
Yu
*a
aKey Laboratory of Marine Sediment and Environmental Geology of State Oceanic Administration, First Institute of Oceanography, State Oceanic Administration, Qingdao, 266061, PR China. E-mail: hjyu@fio.org.cn; Fax: +86-532-84022681
bKey Laboratory of Eco-Chemical Engineering, Ministry of Education, College of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Qingdao, 266042, PR China. E-mail: lifeng@qust.edu.cn
First published on 4th March 2010
Abstract
A new and effective strategy was proposed for the preparation of an organic-inorganic composite matrix by using spherical silica as a supporting core and porous chitosan (CS) hybrid layer as shell, based on sol–gel reaction and simple treatment with ammonia solution. After metal ion loading, immobilized metal affinity adsorbent for protein adsorption was obtained. In the prepared composite matrix, the coating layer was covalently bonded on the supporting silica through polysaccharide incorporated sol–gel process starting from CS and an inorganic precursor γ-glycidoxypropyltrimethoxysiloxane (GPTMS). This siloxane possessed an epoxide group to cross-link amine groups of CS. Scanning electron microscopy investigation showed that the wet phase inversion of CS in ammonia solution endowed the coated CS hybrid layer with a porous surface. X-Ray diffraction investigation revealed significant decrease of CS crystallization, indicating the availability of active amine groups. The as-prepared matrix was also characterized using simultaneous thermogravimetry and differential scanning calorimetry. After loading Cu2+ as pseudo-biospecific ligand, new immobilized metal affinity adsorbent was obtained and its protein adsorption performance was evaluated by batch adsorption experiments using bovine serum albumin (BSA) as a simple model protein. The affinity adsorbent showed fast kinetics and high capability for BSA adsorption. The proposed approach and the prepared matrix showed potential as a platform to conduct bioanalysis.
1. Introduction
Nowadays, the separation and purification of proteins are remarkably concerned in fields of biochemistry, biotechnology and therapeutics. Immobilized metal affinity chromatography (IMAC) has been one of the most effective approaches for analytical and preparative protein separation.1–3 IMAC uses immobilized metal ions as pseudo-biospecific ligands. His-tagged proteins or proteins naturally containing certain functional amino acids can bind to immobilized metal ions. Such affinity binding is the principle for protein adsorption and separation.4–6 Development of a novel matrix with facile functionality for metal ion immobilization and the resulting protein adsorption, therefore, is of great significance.Chitosan (CS), is a natural polysaccharide composed of glucosamine and N-acetylglucosamine residues with 1,4-β-linkage. As a typical biological macromolecule, CS contains a set of unique characteristics including hydrophilicity, nontoxicity, biocompatibility, and physiological inertness. In addition, CS material is a promising IMAC matrix because the high concentration of amino groups in CS chains confers excellent binding capacity with heavy metal ions. Consequently, chelating-ligand coupling process for the preparation of conventional IMAC matrix could be total eliminated. However, pure CS material has unsatisfying mechanical properties. The preparation of a core-shell composite material by coating CS-based layer on preformed particle has been proven as a conventional way to develop CS-based matrix in chromatography.4–10
Organic–inorganic composite materials have recently attracted much attention in both industry and research. The preparation of organic–inorganic composite materials could be divided into two methods. One is the preparation of organic material-supported composite material. The other is the preparation of organic–inorganic composite materials based on hybridization of an organic component with an inorganic source using a sol–gel reaction. Silica has been shown to be a good candidate as supporting material because of its large surface area and excellent mechanical resistance.11,12 The essence of the preparation of silica-supported organic-inorganic composite material is the combination of the functionality of CS and advantages of silica, e.g., large surface area and excellent mechanical resistance. For example, Shi’s group prepared a novel IMAC matrix for protein adsorption by coating CS on silica bead.13 Wu’s group prepared macroporous silica-CS matrix by coating CS on non-porous silica. In their work, polyethylene glycol was used as porogen and epichlorohydrin was used as cross-linking reagent.5 For the preparation of organic–inorganic composite materials based on hybridization on molecular level, sol–gel technology offers simple and convenient methodology.14 Our group prepared porous matrices by incorporation CS into an inorganic network based on cross–linked organic-inorganic hybridization in sol–gel process and molecular imprinting.11,12 In the prepared hybrid matrices, the functional amino groups of CS are covalently attached to the rigid inorganic network using an organosiloxane containing an epoxide ring as precursor.4,15,16 The morphology of the obtained material could be controlled by changing the imprinting molecules in molecular imprinting technology.
This work discloses a new and efficient approach to prepare a new matrix with porous and functional surface. In the prepared matrix, CS and an epoxide-contained siloxane were covalently hybridized in a sol–gel reaction resulting in a CS hybrid layer coated on the supporting spherical silica. The following wet phase inversion by simple treatment with ammonia solution endowed the composite material with a porous surface. Though no imprinting technique was used, the as-prepared adsorbent exhibited a porous surface. After the loading of Cu2+ as an affinity ligand, the obtained material shows promising as a new immobilized metal affinity adsorbent for protein adsorption. The preparation methodology, main characteristic features and application for protein adsorption of the new biomaterial are described and discussed in detail.
2. Experimental
2.1 Instrumentation
Scanning electron microscopy (SEM) images were obtained at 5.0 kV on a Hitachi S-4100 field emission scanning electron microscope (Hitachi, Japan). X-Ray diffraction (XRD) was performed on Siemens D 5005 powder X-ray diffractometer. Simultaneous thermogravimetry and differential scanning calorimeter (TG/DSC) were conducted with a NETZSCH STA 409 thermogravimetric analyzer (Bruker, USA). A TAS-990 atomic absorption spectrophotometer (Beijing Purkinje General Instrument Co., Ltd.) was used to measure the concentration of metal ions in aqueous solution.2.2 Chemicals
Chitosan, with 98% deacetylation and an average molecular weight of 8 × 104 g mol−1 (Yuhuan biomedical corp., China) was used to cast spherical silica (105 μm, Qingdao Ocean Chemical Co., China). All other chemicals used were of analytical grade. Doubly deionized water (DDW) was used throughout this work.2.3 Preparation of the silica supported CS matrix
Silica was heated at 110 °C for 1 h to activate the surface. A 2% CS solution was dissolved in 1 M acetic acid aqueous solution. After stirring for 1 h, a perfectly transparent solution was obtained and 2% GPTMS was added. The mixture was then placed in an ultrasonic bath for 30 min. The activated silica was then added to the CS-GPTMS solution in a flask. The amount of solution coated on silica was controlled at 2 mL per gram silica. The obtained flask was placed in the incubator for surface coating reaction to stay overnight at room temperature. The beads coated with CS were then immersed in a concentrated ammonia solution for 6, 12, 18, and 24 h. Then the resulting material was separated from the concentrated ammonia solution, and washed several times with DDW to the neutral pH. The wet beads were allowed to dry at room temperature to obtain dry product. The dry product was stored in desiccators while not in use. The morphologies of the obtained materials were characterized using scanning electron microscope after gold plating. The thermal and decomposition characteristics of such materials were determined by TG/DSC in the temperature range of 50–850 °C at a heating rate of 10 °C min−1, with air flow at 200 mL min−1.2.4 Preparation of immobilized metal affinity adsorbent by Cu2+ coordination
The immobilized metal affinity adsorbent was prepared by Cu2+ loading. In all batch experiments, 1 g of each type of bead was equilibrated with 10 mL of acetic acid/sodium acetate buffer solution (pH 5.0) containing 2 mg mL−1 Cu2+ in stoppered plastic vials. The mixtures were stirred for 30 min at room temperature. The filtrates were measured to obtain Cu2+ loading capacity by atomic absorption spectrometer. The matrix could be regenerated because Cu2+ immobilized on the adsorbents could be stripped by washing with EDTA at 0.01 M. Afterwards, the matrices were thoroughly washed with DDW and Cu2+ could bind with the regenerated matrix.2.5 Bovine serum albumin (BSA) adsorption test
Binding kinetics of BSA to the prepared immobilized metal affinity adsorbent was performed by adding 300 μL BSA solution (10 mg mL−1) to 1 g adsorbent immersed in 10 mL phosphate buffer solution (PBS) at pH 6.0. After shaking for different times, the content of BSA in the supernatant was measured. Such time-dependent adsorption tests indicate that 50 min is sufficient for adsorption.The effect of pH on the adsorption of BSA was tested by equilibrating the prepared immobilized metal affinity adsorbents (1 g) with 10 mL of the buffer solutions containing 1 mg mL−1 of BSA under different pH conditions. The pH of the buffer was adjusted using 20 mM PBS for pH ranged from 4.0 to 7.5. For comparison, the BSA adsorption to the initial matrix without Cu2+ loading was also determined to investigate the non-specific adsorption.
Adsorption isotherm of BSA was determined by equilibrating different concentration of BSA with the prepared adsorbent to measure the adsorption capacity. 1 g Adsorbent was equilibrated with 10 mL of the PBS buffer solutions at pH 6.0 containing various concentrations of BSA in stoppered plastic vials. The mixtures were stirred for 50 min at room temperature (25 °C).
In all above batch experiments, the mixtures were separated by centrifugation. The amount of BSA adsorbed was calculated by the following eqn (1).
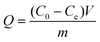 | (1) |
2.6 Determination of BSA
The content of BSA was measured by the method of Bradford.173. Results and discussion
3.1 Preparation and characterization of the silica supported CS composite matrix based on simple treatment with ammonia solution
In this work, a new and effective strategy was proposed for the preparation of new organic-inorganic composite matrix by using spherical silica as supporting core and porous CS hybrid layer as shell based on sol–gel reaction and simple treatment with ammonia solution.Scanning electron microscopy (SEM) was used to investigate the surface morphology of the prepared matrix. Fig. 1 shows the morphology of the supporting silica. As shown, the supporting silica possesses a diameter of about 105 μm. Moreover, the supporting silica exhibited a rigid surface. Fig. 2 provides a comparison between the surface of the hybrid material before and after the treatment with ammonia solution. As seen, the two hybrid materials exhibit different morphologies. It was obvious that a porous surface was obtained after the composite material was treated with ammonia solution. The phenomenon indicated that ammonia solution gave a porous structure for the coated CS hybrid layer. That might be caused by the phase inversion because the excess acid was neutralized by ammonia solution. Moreover, the volatilization of ammonia might result in the porous surface.18
 | ||
| Fig. 1 Scanning electron microscopy image of the supporting silica. | ||
 | ||
| Fig. 2 Scanning electron microscopy image of the CS hybrid layer coated composite matrix before (a) and after (b) treated with ammonia solution. | ||
Fig. 3 shows the XRD pattern of pure CS and the hybrid matrix before (b) and after (c) treatment with ammonia solution. Such material was prepared by casting CS-GPTMS sol–gel solution on a clean glass plate and analyzing the scraped power after treatment. As shown in Fig. 3a, there are two strong peaks in the diffractogram of CS at 2θ = 11.4° and 20.2°. However, when the composite matrix was treated with concentrated ammonia there is no peak for crystalline regions as compared with pure CS (Fig. 3c), indicating significant decrease in the degree of crystallinity. Compared with the hybrid material before treatment with ammonia solution, such decrease was larger. Therefore, it was expected that the organic-inorganic hybridization and the treatment with ammonia solution increase the hydrophilicity and the diffusion properties of CS. As a result of the reduced crystallinity, the availability of functional amine groups and metal binding capacity were improved.8
 | ||
| Fig. 3 X-Ray diffraction patterns of CS (a) and the prepared composite matrix before (b) and after (c) treatment with ammonia solution. | ||
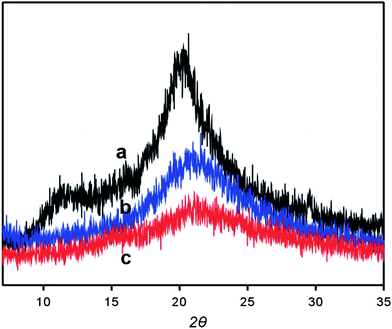
By using TG/DSC, the thermal stability of pure CS and the obtained composite material treated with ammonia solution for 2 h or 12 h was investigated. As shown in Fig. 4, the thermogravimetric curve of the three materials showed two main degradation stages. The first weight loss occurred around 100 °C, which was attributed to the loss of adsorbed water molecules. The second weight loss region (200–400 °C) might be caused by the decomposition of CS in the air flow. For pure CS, a significant weight loss was observed. For the material treated with ammonia solution for 2 h, the loss was small, indicating low CS content on the supporting silica. However, the weight loss was greater for the material that was treated with ammonia solution for 12 h. The phenomenon indicated the strength of the obtained CS hybrid shell might be affected by the treatment time in ammonia solution. Short treatment time resulted in low strength of the obtained CS hybrid shell. As a result, the coated CS layer is easily lost in the process such as washing and so on.
 | ||
| Fig. 4 The TG/DSC analysis of CS and the composite matrix prepared after treatment with ammonia solution for 2 h or 12 h. | ||
3.2 The loading of the affinity ligand on the matrix
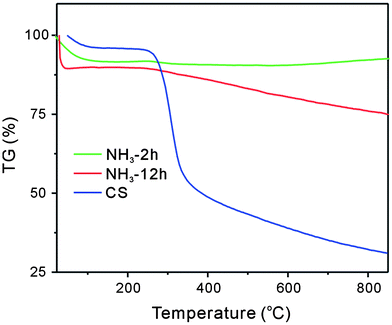
In this work, Cu2+ was chosen as the affinity ligand. Fig. 5 shows the Cu2+ loading capacity of the matrices prepared by treatment with ammonia solution for different durations of time. As seen, the treatment time showed significant effect for the capacity of Cu2+ loading. When the treatment time was not less than 12 h, the Cu2+ loading capacity was similar. The material treated for 18 h showed the highest loading capacity. Since the available functional ligands (–NH2) were used for metal ion binding, the capacity of Cu2+ loading increased when CS grafted on the silica increased. The strength of the CS layer might be low when the treatment was not enough. Even for the material obtained after being treated with ammonia for 6 h, however, the Cu2+ loading capacity was remarkably higher than the blank loading capacity of the bare silica. The phenomenon indicated that the presence of functional CS hybrid shell gave specific binding of Cu2+. The Cu2+ loading capacity was higher than those obtained on CS/sand,13 CS/glass19 and CS/silica5,20 composite materials. | ||
| Fig. 5 Effect of treatment time in ammonia solution on the Cu2+ adsorption capacity of the prepared composite matrix. | ||
3.3 BSA adsorption on the prepared affinity adsorbent
After the loading of Cu2+, the matrix turned into an immobilized metal affinity adsorbent. BSA was chosen as the model protein due to the presence of His on the protein surface.12 The adsorption of protein in IMAC was governed by the coordination of metals with electron-donor ligands exposed on the surface of the proteins. So the effect of pH on BSA adsorption was investigated using adsorbent obtained from the matrix treated with ammonia solution for 12 h. Results are given in Fig. 6. The binding of BSA to the adsorbent increased with the pH, reaching an optimum at pH 6.0. A fairly similar trend had been reported by Wu and our group.5,12 As no copper ions were released under the experimental conditions, it was a clear indication that the ability of protein coordination with immobilized Cu2+ decreased in more acidic conditions. The tendency might be ascribed to the protonation of electron donor group in amino acid residues, such as imidazole nitrogen in His. In the following experiments, PBS at pH 6.0 was chosen as the binding buffer.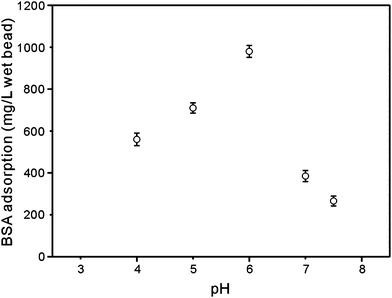 | ||
| Fig. 6 The dependence of BSA adsorption capacity on pH of the adsorption solution. The affinity adsorption was prepared by loading Cu2+ on the composite matrix prepared by treatment in ammonia solution for 12 h. | ||
The effect of the treatment time in ammonia solution on BSA adsorption is given in Fig. 7. As seen, the material treated in ammonia solution for 12 h possessed the highest BSA binding though the highest Cu2+ loading was obtained by material treated for 18 h. As a result, the protein binding might be affected by the surface characteristic and the amount of affinity ligands. Such synthetic effects resulted in different density of affinity ligands. In the following experiments, the matrix treated in ammonia solution for 12 h was chosen for the preparation of affinity adsorbent.
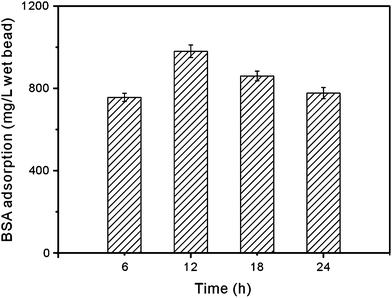 | ||
| Fig. 7 BSA adsorption to the immobilized metal adsorbent treated in ammonia solution for different time. | ||
3.4 Binding kinetics and adsorption isotherm
The kinetic profile of BSA adsorption to the prepared immobilized metal affinity adsorbent was investigated. It can be seen from Fig. 8 that BSA possessed a fast adsorption profile in the initial step with significant specific binding within 10 min.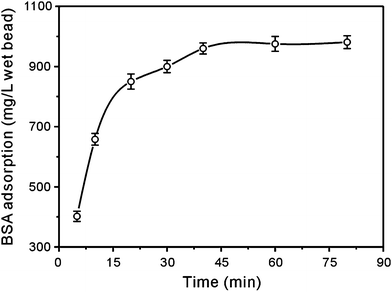 | ||
| Fig. 8 Kinetic curve of BSA adsorption on the immobilized metal affinity adsorbent. | ||
The adsorption isotherm of BSA to the prepared immobilized metal affinity adsorbent was similar to the Langmuir adsorption isotherm. Data shown in Fig. 9 was fitted to the linear form of the Langmuir equation:
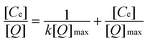
The calculated coefficient k = 4.9 L mol−1 and [Q]max = 980 mg per liter wet bead indicated that the adsorbent had a considerable adsorption capacity and could be used as adsorbent for BSA adsorption from aqueous solution.
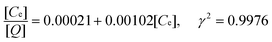
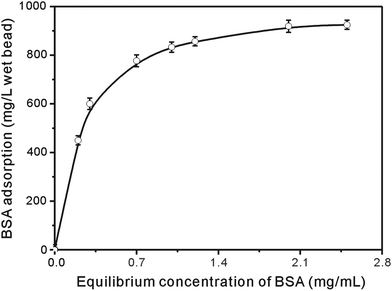 | ||
| Fig. 9 Adsorption isotherm of BSA to the immobilized metal affinity adsorbent at pH 6, T = 25 °C. | ||
4. Conclusions
The successful preparation of the organic-inorganic composite matrix with spherical silica as supporting core and porous CS hybrid as shell demonstrated the feasibility of simple treatment with ammonia solution. The method is simple and effective. Such treatment gave a porous structure for the coated CS layer. The crystallinity of CS was reduced, increasing the availability of amine functional groups and metal binding capacity. The formed affinity adsorbent showed good protein binding capacity and might serve as a platform to produce chromatographic beads. This methodological study and application in the affinity adsorption of protein might be an important field.Acknowledgements
This research was supported by the National Natural Science Foundation of China (Nos. 20775039 and 40602018), the Natural Science Foundation of Shandong Province of China (No. ZR2009BM031), and the Open Foundation of State Key Laboratory of Electroanalytical Chemistry (No. 2009004).References
- M. C. Cheeks, N. Kamal, A. Sorrell, D. Darling, F. Farzaneh and N. K. H. Slater, J. Chromatogr., A, 2009, 1216, 2705 CrossRef CAS.
- S. D. Alexandratos and X. P. Zhu, React. Funct. Polym., 2007, 67, 375 CrossRef CAS.
- F. Le Dévédec, L. Bazinet, A. Furtos, K. Venne, S. Brunet and M. A. Mateescu, J. Chromatogr., A, 2008, 1194, 165 CrossRef CAS.
- F. N. Xi, J. M. Wu and X. F. Lin, J. Chromatogr., A, 2006, 1125, 38 CrossRef CAS.
- F. N. Xi and J. M. Wu, J. Chromatogr., A, 2004, 1057, 41 CrossRef CAS.
- V. K. Mourya and N. N. Inamdar, React. Funct. Polym., 2008, 68, 1013 CrossRef CAS.
- F. Li, W. Chen, C. F. Tang and S. S. Zhang, Talanta, 2009, 77, 1304 CrossRef CAS.
- F. Li, H. Q. Jiang and S. S. Zhang, Talanta, 2007, 71, 1487 CrossRef CAS.
- Y. Xiao and X. H. Zhou, React. Funct. Polym., 2008, 68, 1281 CrossRef CAS.
- F. N. Xi, J. M. Wu, Z. S. Jia and X. F. Lin, Process Biochem., 2005, 40, 2833 CrossRef CAS.
- F. Li, P. Du, W. Chen and S. S. Zhang, Anal. Chim. Acta, 2007, 585, 211 CrossRef CAS.
- F. Li, X. M. Li and S. S. Zhang, J. Chromatogr., A, 2006, 1129, 223 CrossRef CAS.
- Q. H. Shi, Y. Tian, X. Y. Dong, S. Bai and Y. Sun, Biochem. Eng. J., 2003, 16, 317 CrossRef CAS.
- J. H. Shin, S. W. Weinman and M. H. Schoenfisch, Anal. Chem., 2005, 77, 3494 CrossRef CAS.
- Y. L. Liu, Y. H. Su and J. Y. Lai, Polymer, 2004, 45, 6831 CrossRef CAS.
- Y. Shirosaki, K. Tsuru, S. Hayakawa, A. Osaka, M. A. Lopes, J. D. Santos and M. H. Fernandes, Biomaterials, 2005, 26, 485 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248 CrossRef CAS.
- T. Y. Guo, Y. Q. Xia, G. J. Hao, M. D. Song and B. H. Zhang, Biomaterials, 2004, 25, 5905 CrossRef CAS.
- D. C. Nash, G. E. McCreath and H. A. Chase, J. Chromatogr., A, 1997, 758, 53 CrossRef CAS.
- M. W. Wan, I. G. Petrisor, H. T. Lai, D. Kim and T. F. Yen, Carbohydr. Polym., 2004, 55, 249 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |