Improving the depth of coverage in membrane proteomic studies through the use of lipid-based protein immobilization technology in parallel with methanol-facilitated solubilisation†
Neerav D.
Padliya
*,
Mohit B.
Bhatia
,
Wolfgang T.
Hofgärtner
and
Robert J.
Hariri
Celgene Cellular Therapeutics, Division of Celgene Corporation, 7 Powder Horn Drive, Warren, NJ 07059, USA. E-mail: npadliya@celgene.com; Fax: +1 (732)-652-5701; Tel: +1 (732)-564-3454
First published on 23rd February 2010
Abstract
Lipid-based protein immobilization (LPI) technology is a platform recently developed to facilitate shotgun membrane proteomic studies based on a nanotechnology framework. Proteoliposomes are generated from a membraneprotein preparation. These proteoliposomes are immobilized onto an LPI chip and then subjected to proteolysis. The proteolytic peptides are then subjected to LC/MS analysis after fractionation by SCX chromatography. The focus of this study was to evaluate how the depth of coverage of the membrane proteome of a particular cell type varied as a function of the sample preparation method used. Human dermal fibroblasts (hDFs) and human bone marrow mesenchymal stem cells (BM-hMSCs) were subjected to membrane proteomic studies using two different sample preparation methods: LPI technology and methanol-facilitated solubilisation. The number of membraneproteins that could be identified from hDFs and BM-hMSCs using LC/MS was greater using LPI technology than it was using methanol-facilitated solubilisation. However, the number of membraneprotein identifications that could be made for both hDFs and BM-hMSCs increased by ∼50% when both sample preparation methods were used in parallel and the MS/MS data was convolved together. Therefore, LPI technology is a very useful technology for high-throughput shotgun membrane proteomic studies. However, in order to maximize the depth of membrane proteome coverage that can be attained for a given cell type, it is necessary to use multiple sample preparation methods in concert.
Introduction
In the rapidly advancing field of stem cell research, mass spectrometry (MS)-based proteomic technologies have taken on a prominent role. MS-based proteomics is making it possible to shed light into the mechanism of action of stem cells by enabling the elucidation of cellsignaling pathways,1 the identification of secreted proteins from conditioned media,2 and the comprehensive phenotyping of the membrane proteome.3 A number of stem cell-based therapeutic approaches exploit the immunomodulatory capabilities of certain stem cells for the treatment of diseases such as Crohn's Disease,4 scleroderma,5 lupus6 and other inflammatory diseases. It is well known that many of the proteins expressed at the cell surface of stem cells play an important immunoregulatory role.One of the major advantages of using mass spectrometry to characterize the membrane proteome of stem cells is that mass spectrometry can identify proteins and the post-translational isoforms of those proteins without the need for protein-specific reagents such as antibodies. Foster and colleagues capitalized on this advantage of mass spectrometry to shed light into the process by which mesenchymal stem cells undergo osteoblast differentiation in a quantitative proteomics study that involved SILAClabeling.3 Due to the hydrophobicity and microheterogeneity of membraneproteins, the characterization of this class of proteins by LC/MS methods continues to remain a major technical challenge. As a result, a number of creative approaches to comprehensively profile the membrane proteome have been published in the literature.7–9 By exploiting the use of multiple proteases in tandem along with optimized digestionbuffer conditions, Dormeyer and colleagues10 used tandem mass spectrometry to identify 237 plasma membrane specific proteins from the human embryonic stem cell line, HUES-7. Their rigorous experimental work led to the identification of plasma membraneprotein markers that can be used to distinguish embryonic stem cells from embryonal carcinoma cells which will likely contribute to an improved understanding of the safe therapeutic use of stem cells. In a comprehensive review, Wu and Yates11 discussed the use of various detergents, organic solvents or organic acids in the presence of trypsin or cyanogen bromide to solubilise and digest membraneproteins prior to LC/MS characterization in high-throughput shotgun membrane proteomic studies. Lu et al.12 used a tube-gel digestion approach to identify 178 membraneproteins from prostate cancer cells making use of only a single digestion step and 2.5 h of LC/MS time.
One altogether different approach taken towards membraneprotein characterization by LC/MS that capitalizes on recent advances in nanotechnology was described by Nyblom et. al.13 In this paper, a recombinant membraneprotein, human aquaporin 1 (hAQP1), was characterized by generating proteoliposomes through a tip-sonication process followed by immobilizing these proteoliposomes generated from hAQP1 onto a solid surface exploiting a nanotechnology framework referred to as lipid-based protein immobilization (LPI™) technology (Nanoxis AB, Gothenburg, Sweden). During the fabrication process of a chip based on LPI™ technology, membraneophilic surfaces are created through the plasma treatment of gold and aluminium oxide. LPI™ technology exploits the affinity of the lipid components of a proteoliposome for the membraneophilic surfaces that comprise an LPI™ chip that were described earlier.13 It is this affinity that makes it possible to rapidly exchange solution conditions without disturbing the immobilized membraneproteins. The ability to immobilize membraneproteins onto a solid surface is very useful because it allows the use of multiple proteases as part of the sample preparation process prior to LC/MS. Proteases such as trypsin require basic solution conditions while proteases such as pepsin require acidic solution conditions. The LPI™ chip facilitates the rapid exchange of solution conditions and makes it possible to digest the same membraneprotein preparation using multiple proteases and solution conditions in tandem. This is often required in order to comprehensively characterize the membrane proteome of a particular cell type using tandem mass spectrometry methods. The LPI™ chip also significantly enhances the efficiency of the proteolysis process as a protease can rapidly access the proteoliposomes immobilized on the two membraneophilic surfaces. Further, the LPI™ process circumvents the need to use detergents such as SDS which can significantly complicate downstreamLC/MS analysis, even after significant sample clean-up. In a recent study by Bauer et al.,14 the use of LPI technology was extended from the characterization of a single recombinant membraneprotein to an entire membrane proteome.
We were interested in determining whether there is any significant advantage to using lipid-based protein immobilization (LPI) technology as part of the sample preparation step in membrane proteomic studies with respect to conventional membrane proteomic sample preparation methods such as those discussed earlier.10–12 Our primary criterion for evaluation was the depth of membrane proteome coverage that could be attained using both of the aforementioned sample preparation methods. In order to investigate this, we characterized a human bone marrow mesenchymal stem cell line (BM-hMSC) and a human dermal fibroblast (hDF) cell line using two workflows; one workflow based on LPI™ technology compared to a conventional workflow based on methanol-facilitated solubilisation described in detail by Blonder et. al.15 When characterizing both BM-hMSCs and hDFs, we identified plasma membrane sub-proteomes of significant size that were exclusive to either the LPI™ workflow or the methanol-facilitated solubilisation workflow. Therefore, LPI™ technology and methanol-facilitated solubilisation can be used in parallel to significantly enhance the depth of coverage that can be attained in large-scale membrane proteomic studies.
Experimental
Cell culture
Human bone marrow mesenchymal cells (BM-hMSCs) and normal human dermal fibroblasts (hDFs) were acquired from ScienCell Research Laboratories (Carlsbad, CA). Each of the two cell types was cultured using growth media provided by ScienCell Research Laboratories as per the supplier's instructions. Cells were harvested from T-flasks by trypsinization. After centrifugation, the cell pellet was washed twice with Hank's balanced salt solution (HBSS) buffer, and then flash frozen to maintain the cell pellet prior to proteomic analysis.Sample preparation
Strong-cation exchange (SCX) chromatography
Tryptic peptides prepared using either the methanol-facilitated solubilisation workflow or the LPI™ workflow were dried down in a Speed-Vac system, reconstituted in 1 mL of 2% acetonitrile, 0.1% formic acid and then desalted using a Waters HLB™ solid-phase extraction (SPE) cartridge (Waters Corporation, Milford, MA). After elution from the SPE cartridge, the tryptic peptides were dried again in a Speed-Vac system and then reconstituted in 500 μL of a 10 mM ammonium formate, 45% acetonitrile (ACN), pH 2.7 buffer (Buffer A). Tryptic peptides were then injected onto a 1.0 mm × 150 mm polySULFOETHYL A™ column (PolyLC, Inc., Columbia, MD) and separated by employing an increasing two-step linear gradient from 100% Buffer A to 100% Buffer B (500 mM ammonium formate, 45% acetonitrile, pH 2.7) while monitoring 280 nm absorbance over the course of 50 min. One salt fraction was collected every minute over the course of the 50 min SCX run and then pooled together to give rise to 16 SCX fractions. Each SCX fraction was dried down in a Speed-Vac system and then reconstituted in 15 μL of 2% ACN, 0.1% formic acid prior to LC/MS analysis.Liquid chromatography-mass spectrometry analysis
Tryptic peptides were injected onto a Halo™ C18 column (0.2 × 150 mm, 2.7 μm) (Michrom Bioresources, Auburn, CA) and separated using an increasing acetonitrile gradient. Peptides were ionized using an axial desolvation vacuum-assisted nanocapillary electrospray (ADVANCE) ionization source (Michrom Bioresources, Auburn, CA) and then introduced into an LTQ linear ion trap mass spectrometer (Thermo Fisher Scientific, San Jose, CA). A parent ion scan was performed over the range 400–2000 m/z. Automated peak recognition, dynamic exclusion, and MS/MS ion scanning of the top five most intense parent ions were controlled by Xcalibur 2.0.7 (Thermo Fisher Scientific, San Jose, CA).Bioinformatics
Each of the 16 .RAW files generated from a single LC/MS experiment was searched against the SWISSPROT database using an implementation of the Sequest algorithm on a Sorcerer Solo™ workstation (Sage-N Research, Milpitas, CA). Carbamidomethylation was used as a static modification, the oxidation of methionine was used as a differential modification and the peptide mass tolerance was set to 1.2 amu. Protein identifications were parsed using Scaffold 2.0 (Proteome Software, Portland, OR). Protein identifications were only accepted if both the peptide probability and protein probability thresholds exceeded 95% and if two unique peptides were identified. All protein identifications meeting the aforementioned criteria were exported into the Ingenuity Pathway Analysis (IPA) software (Ingenuity Systems, Redwood City, CA). The enriched dataset feature in IPA was used to classify all protein identifications according to subcellular location.Results and discussion
The purpose of this study was to evaluate the feasibility of using lipid-based protein immobilization (LPI™) technology to facilitate high-throughput shotgun membrane proteomic studies. Therefore, we used a sample preparation method based on LPI™ technology as part of a mass spectrometry-based workflow to characterize the membrane proteome of two cell lines, BM-hMSCs and hDFs. In particular, we were most interested in determining the depth of membrane proteome coverage that could be attained when LPI™ technology was used as part of a workflow to characterize BM-hMSCs and hDFs by LC/MS. Both of these two cell lines were also characterized using an alternate sample preparation method, methanol-facilitated solubilisation, a sample preparation method that has been commonly used in high-throughput shotgun membrane proteomic studies.15 This made it possible to directly compare the influence of these two different sample preparation methods on the depth of membrane proteome coverage that could be attained for two different mammalian cell lines, hDFs and BM-hMSCs.Both sample preparation workflows (LPI or methanol-facilitated solubilisation) involved the fractionation of the membraneprotein tryptic digest (hDF or BM-hMSC) by offline-SCX chromatography (Fig. 1). In each biological replicate, a total of 16 SCX fractions were collected from each of the four treatment conditions studied in this investigation. All 16 SCX fractions were later subjected to LC/MS analysis after sample clean-up. For each of the four treatment conditions outlined in Fig. 1, LC/MS datasets were collected from a total of three biological replicates. All of the .RAW files collected from each one of the four treatment conditions were searched against the SWISSPROT database using an implementation of the SEQUEST algorithm on a Sorcerer Solo™ workstation. Scaffold 2.0 software was used to assess the confidence of protein identifications made by database searching. Protein identifications with protein probability above 95% were retained for further analysis. Using the Ingenuity Pathway Analysis (IPA) software, we were able to rapidly classify all membraneproteins exported from Scaffold 2.0 according to their subcellular location and focus our attention specifically on the plasma membraneproteins. An average number of plasma membraneprotein identifications was calculated (rounded to the nearest whole number) for each of the four treatment conditions (Fig. 1) and referred to in subsequent comparisons.
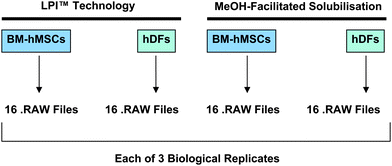 | ||
| Fig. 1 Membrane protein tryptic digests derived from BM-hMSCs and hDFs were prepared using sample preparation methods based on LPI™ technology and MeOH-facilitated solubilisation. Each of the four tryptic digests were subjected to offline SCXfractionation. 16 SCX fractions were collected from each of the four tryptic digests. Each of the 16 SCX fractions were subjected to LC/MS analysis giving rise to 16 .RAW files. | ||
Using a sample preparation method based on LPI™ technology, we identified 233 plasma membraneproteins from BM-hMSCs using tandem mass spectrometry (Fig. 2A). When this cell line was characterized by tandem mass spectrometry using a sample preparation method based on methanol-facilitated solubilisation, a total of 226 plasma membraneproteins were identified. Although the total number of plasma membraneproteins identified by BM-hMSCs was very comparable, the overlap between the specific membraneproteins identified by the two sample preparation was surprisingly less than 30%. The total number of non-redundant proteins identified from BM-hMSCs using these two sample preparation methods in parallel was 354, an increase of 51.9% with respect to the number of membraneproteins that could be identified using a sample preparation method based on LPI™ technology alone.
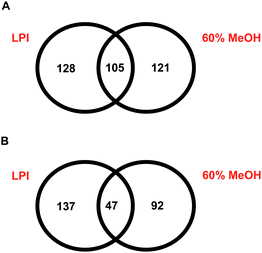 | ||
| Fig. 2 A: Identification of BM-hMSCplasma membraneproteins using LPI™ technology and methanol-facilitated solubilisation. B: Identification of hDFplasma membraneproteins using LPI™ technology and methanol-facilitated solubilisation. | ||
When hDFs were subjected to mass spectrometry-based membrane proteomic characterization using two different sample preparation methods in parallel (LPI™ and methanol-facilitated solubilisation), we found that the overlap between the membraneproteins identified using these two workflows was only 17% (Fig. 2B). In this particular case, 184 plasma membraneproteins were identified from hDFs using the LPI™ sample preparation method while only 139 membraneproteins were identified from hDFs using the methanol-facilitated solubilisation method. However, using these two sample preparation methods in parallel led to the identification of 276 plasma membraneproteins, an increase of 50% with respect to the number of membraneproteins that could be identified using a sample preparation method based on LPI™ technology alone.
The results discussed earlier and presented in Fig. 2 will naturally prompt one to ask, “why is the overlap between plasma membraneprotein identifications made using the LPI™ technology method versus those made with the methanol-facilitated solubilisation method not as large as one would expect?” There are three major differences between how membraneprotein-derived tryptic peptides were generated using LPI™ technology method with respect to the methanol-facilitated solubilisation method. The first difference is that in the methanol-facilitated solubilisation method, the membraneprotein pellet was tip-sonicated only for 1–2 min. On the other hand, when preparing membraneprotein samples using the LPI™ method, tip sonication was carried out for 60 min. As the length of the tip-sonication cycle is increased, the average membraneprotein fragment size decreases substantially as one would expect (unpublished results from fluorescence microscopy). It is very likely that the average membraneprotein fragment size will affect the nature of the tryptic peptides that are generated following digestion as the specific amino acid residues that will be most susceptible to attack by trypsin will be different in the two cases.14 The second difference between these two sample preparation methods is that in LPI™ method, the tip-sonication process is done under aqueous conditions (with the inclusion of 50 mM ammonium bicarbonate) while in the methanol-facilitated solubilisation method, the tip-sonication process takes place under 60% methanol solution conditions. The solution conditions that are employed during the tip-sonication process likely have an effect on the conformation of the membraneprotein fragments that result. These conformational differences can likely account for differences in the nature of tryptic peptides that result due to differences in susceptibility to proteolysis by trypsin. For example, Russell and colleagues16 reported major differences in tryptic digest profiles that were obtained when a given protein was digested in an organic solvent as opposed to an aqueous solvent. The third difference between the two sample preparation methods is that the LPI™ method employs immobilization of proteoliposomes prior to digestion while the methanol-facilitated solubilisation method does not employ immobilization. If we assume that the proteoliposomes generated through the tip-sonication process are not perfectly homogeneous on the periphery, then they will likely have regions that have a stronger affinity towards the membraneophilic surfaces of the LPI™ chip than other regions. This would explain how the immobilization process can affect the nature of the proteolytic peptides that are generated following proteolysis. Therefore, it is not possible to answer the original question regarding the degree of overlap in plasma membraneprotein identifications made using these two sample preparation methods without doing further experiments to isolate the effects of each one of the three factors that were discussed earlier. However, we plan to investigate these effects in a future study.
It is interesting to note that some of the classic phenotypic markers for BM-hMSCs could be identified irrespective of which of the two sample preparation methods (methanol-facilitated solubilisation or LPI™ technology) was used prior to LC/MS analysis of the membraneprotein digest (Table 1). Some of these phenotypic markers included CD44, CD63, CD73, CD95 and CD105. Consistent with the CD34- CD45- phenotype of BM-hMSCs, these membraneproteins were not identified in any one of the LC/MS experiments that were performed with BM-hMSCs. However, a significant fraction of the membraneproteins that are known to be of functional significance in BM-hMSCs could only be identified using a single sample preparation method prior to LC/MS analysis (Table 1). The following membraneproteins are some examples of membraneproteins that could only be identified from BM-hMSCs using a sample preparation method based on LPI™ technology: caveolin 2, fibroblast growth factor receptor 1 (FGF-1), fibroblast growth factor receptor 3 (FGF-3), flotillin 1, hyaluronan synthase 1, insulin-like growth factor 2 receptor, IL-1 receptor, lysosomal-associated membraneprotein 2 (CD107b), vitronectin receptor (CD51), prostaglandin F2 receptor negative regulator, transforming growth factor (TGF), beta receptor II, and vascular cell adhesion molecule 1 (CD106). Hyaluronan synthase 1 is a membraneprotein that synthesizes hyaluronic acid, a major extracellular matrix (ECM) component that plays an important role in the wound healing process, an important therapeutic application of BM-hMSCs.17, 18 FGF receptors 1 and 3, IL-1 receptor, TGF, beta receptor II and CD107b are examples of proteins that are known to have a very important role in terms of immunomodulation, another important application of BM-hMSCs.19, 20 Yet, none of these membraneprotein identifications would have been made had we circumvented the use of LPI™ technology prior to LC/MS analysis. Likewise, a number of membraneproteins of functional significance could only be identified from BM-hMSCs using the methanol-facilitated solubilisation sample preparation method. Examples of such proteins include: activin A receptor, brain-specific angiogenesis inhibitor 3, CD29, CD50, CD74, CD81, cholecystokinin Breceptor, glucagon-like peptide 1 receptor, IL-10 receptor beta, IL-13 receptor alpha 1, IL-6signal transducer, and neural cell adhesion molecule 1 (NCAM-1). Some of these aforementioned proteins are involved in BM-hMSC processes such as osteogenesis21, 22 and immunomodulation.19
| a In Table 1 and Table 2, a red shaded gene symbol indicates that the membraneprotein was only observed using methanol-facilitated solubilisation. A blue shaded gene symbol, indicates that the membraneprotein was only observed using lipid-based protein immobilization technology. A white shaded gene symbol indicates that the membraneprotein was observed using both of the aforementioned sample preparation method. The same color coding scheme has been used for Supplementary Tables 1 and 2†. |
|---|
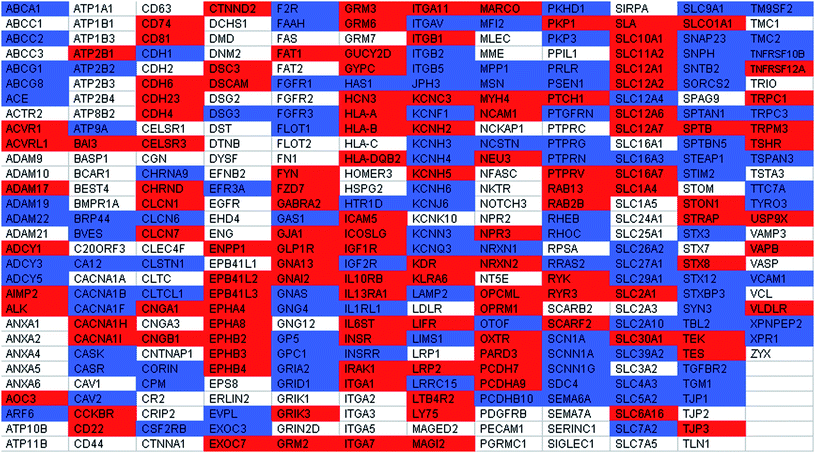
|
Likewise, it was apparent that there were major benefits of using sample preparation methods based on both LPI™ technology and methanol-facilitated solubilisation to characterize membraneprotein tryptic digests derived from hDFs (Table 2). Some examples of membraneproteins with functional relevance to hDFs that were identified using only the sample preparation method based on LPI™ technology were: advanced glycosylation end product-specific receptor (AGER), AXLreceptortyrosinekinase, CD26, CD39, CD48, CD63, CD107b, CD204, CD223, epidermal growth factor receptor pathway substrate 8, flotillin 1, flotillin 2, integrin alpha 8, neuroligin 2, and platelet-derived growth factor receptor, beta polypeptide. On the other hand, there were a number of functionally relevant membraneproteins that were only identified from hDFs using a methanol-facilitated solubilisation workflow such as: adiponectin receptor 1, CD5, CD49c, CD49f, colony stimulating factor 1 receptor, fibroblast growth factor receptor 4 (FGF-4), GDNF family receptor alpha 2, IL-12 receptor beta 1, IL-6signal transducer, and killer cell lectin-like receptor.
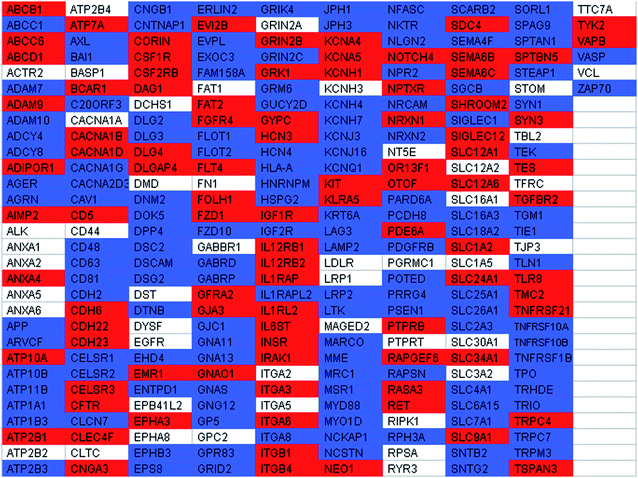
|
In cell therapy research, one of the key reasons for performing a membrane proteomic study of a novel cell line is to gain some insight into the possible mechanism of action of the cell line under investigation. It is quite clear that using sample preparation methods based on both LPI technology and methanol-facilitated solubilisation in parallel significantly enhanced the depth of membrane proteome coverage in studies involving both BM-hMSCs and hDFs, thus improving the likelihood of identifying membraneproteins that actually do play a role in the mechanism of action of these two cell types. It is clear that lipid-based protein immobilization technology has considerable value in terms of facilitating shotgun membrane proteomic studies. Further, it is very likely that even greater depth of membrane proteome coverage can be obtained by using LPI™ technology to facilitate the digestion of membraneproteins with multiple proteases in tandem due to its capability to facilitate the rapid exchange of solution conditions. In addition to experimenting with multiple proteases in tandem, one could experiment with the use of multiple detergents and/or solvent systems prior to digestion. Due to the challenging technical nature of characterizing membraneproteins by mass spectrometry, it is very likely that one will need to use multiple sample preparation conditions in parallel in order to comprehensively profile the membrane proteome of a particular cell type of interest and to maximize the likelihood of identifying membraneproteins.
Conclusions
Lipid-based protein immobilization (LPI) technology can be used as part of a feasible workflow for shotgun membrane proteomic studies. The use of LPI technology in the sample preparation step led to the identification of a greater number of membraneproteins from hDFs and BM-hMSCs than the use of the methanol-facilitated solubilisation sample preparation method. However, when both sample preparation methods were used in parallel to characterize hDFs and BM-hMSCs, the number of protein identifications increased by ∼50% with respect to the number of membraneproteins that could be identified using only LPI technology alone. In order to comprehensively characterize the membrane proteome of mammalian cells, it is necessary to use multiple sample preparation methods in parallel due to the technically challenging nature of characterizing membraneproteins by LC/MS.List of abbreviations
| ACN | Acetonitrile |
| BM-hMSCs | Human bone marrow mesenchymal stem cells |
| HBSS | Hank's balanced salt solution |
| hDFs | Human dermal fibroblasts |
| IPA | Ingenuity Pathways Analysis |
| LPI | Lipid-based protein immobilization |
| MeOH | Methanol |
| SCX | Strong-cation exchange |
| SPE | Solid-phase extraction |
| TCEP | Tris(2-carboxyethyl)phosphine |
Acknowledgements
N.D.P. would like to thank Dr Christine Tavano, Ingenuity Systems for valuable discussions pertaining to the use of Ingenuity Pathway Analysis software for the analysis of proteomic datasets. N.D.P. is grateful to Eric Law, Jennifer Paredes and Tiffany Reddin for sharing their expertise regarding the culture of human bone marrow mesenchymal stem cells and human dermal fibroblasts. We would also like to thank Dr Marian Guelakis, Dr Xiaokui Zhang, Dr James Edinger and Dr Stewart Abbot for helpful comments while editing the manuscript.References
- I. Kratchmarova, B. Blagoev, M. Haack-Sorensen, M. Kassem and M. Mann, Science, 2005, 308, 1472 CrossRef CAS.
- S. K. Sze, D. P. V. de Kleijn, R. C. Lai, E. K. W. Tan, H. Zhao, K. S. Yeo, T. Y. Low, Q. Lian, C. N. Lee, W. Mitchell, R. M. El Oakley and S.-K. Lim, Mol. Cell. Proteomics, 2007, 6, 1680 CrossRef CAS.
- L. J. Foster, P. A. Zeemann, C. Li, M. Mann, O. N. Jensen and M. Kassem, Stem Cells, 2005, 23, 1367–77 Search PubMed.
- Y. Oyama, R. M. Craig, A. E. Traynor, K. Quigley, L. Statkute, A. Halverson, M. Brush, L. Verda, B. Kowalska, N. Krosnjar, M. Kletzel, P. F. Whitington and R. K. Burt, Gastroenterology, 2005, 128, 552 CrossRef.
- J. M. van Laar, D. Farge and A. Tyndall, Ann. Rheum. Dis., 2005, 64, 1515 CrossRef CAS.
- A. E. Traynor, W. G. Barr, R. M. Rosa, J. Rodriguez, Y. Oyama, S. Baker, M. Brush and R. K. Burt, Arthritis Rheum., 2002, 46, 2917 CrossRef.
- A. R. Blackler, A. E. Speers, M. S. Ladinsky and C. C. Wu, J. Proteome Res., 2008, 7, 3028 CrossRef CAS.
- A. E. Speers and C. C. Wu, Chem. Rev., 2007, 107, 3687 CrossRef CAS.
- A. E. Speers, A. R. Blackler and C. C. Wu, Anal. Chem., 2007, 79, 4613 CrossRef CAS.
- W. Dormeyer, D. van Hoof, S. R. Braam, A. J. Heck, C. L. Mummery and J. Krijgsveld, J. Proteome Res., 2008, 7, 2936 CrossRef CAS.
- C. C. Wu, M. J. MacCoss, K. E. Howell and J. R. Yates 3rd., Nat. Biotechnol., 2003, 21, 532 CrossRef CAS.
- X. Lu and H. Zhu, Mol. Cell. Proteomics, 2005, 4, 1948 CrossRef CAS.
- M. Nyblom, F. Oberg, K. Lindkvist-Petersson, K. Hallgren, H. Findlay, J. Wikström, A. Karlsson, O. Hansson, P. J. Booth, R. M. Bill, R. Neutze and K. Hedfalk, Protein Expression Purif., 2005, 56, 552.
- B. Bauer, M. Davidson and O. Orwar, Angew. Chem., Int. Ed., 2009, 48, 1656 CrossRef CAS.
- J. Blonder, K. C. Chan, H. J. Issaq and T. D. Veenstra, Nat. Protoc., 2006, 1, 2784 Search PubMed.
- W. K. Russell, Z. Y. Park and D. H. Russell, Anal. Chem., 2001, 73, 2682 CrossRef CAS.
- H. Li, X. Fu, Y. Ouyang, C. Cai, J. Wang and T. Sun, Cell Tissue Res., 2006, 326, 725 CrossRef CAS.
- X. Fu, L. Fang, X. Li, B. Cheng and Z. Sheng, Wound Repair Regener., 2009, 17, 325 Search PubMed.
- S. Aggarwal and M. F. Pittenger, Blood, 2005, 105, 1815 CrossRef CAS.
- A. Uccelli, V. Pistoia and L. Moretta, Trends Immunol., 2007, 28, 219 CrossRef CAS.
- M. F. Pittenger, A. M. Mackay, S. C. Beck, R. K. Jaiswal, R. Douglas, J. D. Mosca, M. A. Moorman, D. W. Simonetti, S. Craig and D. R. Marshak, Science, 1999, 284, 143 CrossRef CAS.
- G. D'ippolito, P. C. Schiller, C. Ricordi, B. A. Roos and G. A. Howard., J. Bone Miner. Res., 1999, 14, 1115 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Plasma membraneprotein identifications from human bone marrow mesenchymal stem cells (Table S1) and human dermal fibroblasts (Table S2). See DOI: 10.1039/b9ay00267g |
| This journal is © The Royal Society of Chemistry 2010 |