Molecularly imprinted polymer stir bar sorption extraction and electrospray ionization tandem mass spectrometry for determination of 2-aminothiazoline-4-carboxylic acid as a marker for cyanide exposure in forensic urine analysis
Randy
Jackson
a,
Ilona
Petrikovics
b,
Edward P. C.
Lai
c and
Jorn C. C.
Yu
*d
aThe Master of Science in Forensic Science Program, College of Criminal Justice, Sam Houston State University, Huntsville, TX 77341
bDepartment of Chemistry, Sam Houston State University, Huntsville, TX 77341, USA
cOttawa-Carleton Chemistry Institute, Department of Chemistry, Carleton University, Ottawa, ON K1S 5B6, Canada
dThe Master of Science in Forensic Science Program, College of Criminal Justice, Sam Houston State University, Huntsville, TX 77341, USA. E-mail: jornyu@shsu.edu
First published on 17th March 2010
Abstract
In forensic casework, a stable and quantifiable marker is desirable for the determination of cyanide poisoning in biological fluids. 2-Aminothiazoline-4-carboxylic acid (ATCA) is a chemically stable urinary metabolite of cyanide that has been considered to be a reliable biological marker for cyanide exposure. However, endogenous ATCA is always present in low quantity originating from either dietary intake of cyanide or from normal metabolism of amino acids. A selective and sensitive analytical method is needed to determine the endogenous level of ATCA in order to identify cyanide poisoning. The objective of this research was to prepare molecularly imprinted polymers (MIPs) on the surface of a silica stir bar for molecularly imprinted stir bar sorption extraction (MISBSE). Under optimal extraction conditions, the MISBSE could selectively preconcentrate ATCA from urine samples. The binding capacity of one MISBSE stir bar for ATCA was determined to be 35 ± 3 ng (n = 3). Combining MISBSE with electrospray ionization tandem mass spectrometry (ESI/MS/MS), ATCA was detected without derivatization at the 400 ng/mL concentration level. This new strategy of MISBSE-ESI/MS/MS enhanced the selectivity and sensitivity for the detection of ACTA in urine samples.
1. Introduction
The presence of cyanide in an individual can be due to a number of reasons, such as the normal metabolism of amino acids, inhalation of smoke (from cigarette or fire), ingestion of food, exposure to cyanide in a criminal act, or invasion of chemical warfare agents (CWA) during a terrorist attack.1 For forensic casework, a stable and quantifiable marker is needed to determine an accurate level of cyanide poisoning. There are four major markers in blood and urine: the cyanide ion (CN−), thiocyanate (SCN−), 2-aminothiazoline-4-carboxylic acid (ATCA) and its tautomer 2-imino-2-thiazolidine-4-carboxylic acid (ITCA).2 Cyanide and thiocyanate both have their disadvantages as a marker for cyanide exposure. Under physiological conditions cyanide is present as HCN (pKa = 9.2) and can be eliminated from the blood stream within 20 min of exposure.3,4 The deceased would have to be found within 20 min, which is highly unlikely in forensic casework. As for thiocyanate, the major cyanide metabolite found in blood,5 cyanide metabolism is not the only mode of its formation.6,7 The formation of thiocyante is catalyzed by the mitochondrial sulfurtransferase enzyme, rhodanese (EC 2.8.1.1), in the presence of a sulfur donor.8 When chronic administration of cyanide depletes sulfur donors, the formation of thiocyanate is limited and other alternative pathways such as ATCA formation become more important.9 Another important condition that may shift cyanide metabolism in favor of ATCA is the acidosis that accompanies cyanide intoxication. Since the optimal pH for rhodanese is 8.5, when the bodily pH becomes acidic, the formation of ATCA may become the major pathway of cyanide metabolism.3The use of ATCA as a marker for cyanide poisoning is promising due to its stability at ambient, as well as freezing, temperatures,10 and its production is directly related to cyanide exposure. A sensitive gas chromatographic method has been developed by Logue et al. to increase the ability to detect ATCA.2,11 Quantification of the derivatized ATCA was accomplished on a gas chromatograph with a mass selective detector. Good reproducibility and limit of detection (25 ng/mL) were reported. However, the use of solid phase extraction by ion exchange columns and derivatization involved lengthy steps that pose many contamination risks and health issues arising from the derivatization process. A better extraction and detection method is needed to further advance the research on ATCA regarding its normal endogenous level of production.
Molecular imprinting is a process of making molecular imprints within a polymer network.12 The product of molecular imprinting is a molecularly imprinted polymer (MIP). Molecular imprinting techniques have been utilized for a wide variety of applications, including in chromatography,13 immunoassay14 and sensor devices,15 where the MIP functions as a recognition element. For the purpose of this research, the MIP was synthesized on the surface of a stir bar to serve as a selective sorbent for solid phase extraction. As stir bar sorptive extraction (SBSE) has commonly been used to extract trace amount of organic compounds in aqueous systems,16 combining MIP and SBSE to make it a molecularly imprinted stir bar sorption extraction (MISBSE) is expected to be able to pre-concentrate low concentrations of ATCA in a complex matrix. In this work, different from using MIP films for MISBSE,17 the monomer is polymerized from an active compound that is covalently bonded to an inorganic surface.18,19 After MISBSE, the concentrated analyte can then be injected on the instrument for detection, in this work, by flow injection analysis coupled with an electrospray ionization tandem mass spectrometer (FIA-ESI/MS/MS). Since MIP is a valuable tool for selective pre-concentration to separate polar analytes from complex sample matrices, this technique is of general interest for toxicological analysis. In addition, the combination of MIP and mass selective detection by multiple reaction monitoring is also a promising approach.
2. Experimental
2.1 Materials and equipment
Melting point silica capillary tubes (2 mm outside diameter, and 1.5 mm internal diameter) were purchased from Electrothermal (Rochford, Essex, U.K.). Hex screws (0.080 x ½′′, Fastenal, Huntsville, TX) and matching bolts (McMaster-Carr, Santa Fe Springs, CA) were purchased for their magnetic properties. Analytical grade methacrylic acid (MAA) and methane sulfonic acid were purchased from Mallinckrodt (Phillipsburg, NJ). Ethylene glycol diglycidyl ether (EGDE, TCI America, Portland, OR), ammonium persulfate (J. T. Baker, Austin, TX), and γ-(methacryloyloxy)propyl trimethoxysilane (MPS, Alfa Aesar, Ward Hill, MA) were also analytical grade. The 2-aminothiazoline-4-carboxylic acid (ATCA) standard was purchased from Chem Impex International (Wood Dale, IL), and another quantity of ATCA standard was provided as a gift by Dr. Brian Logue (Department of Chemistry and Biochemistry, South Dakota State University, Brooking, SD), and it was synthesized as described by Nagasawa et al.21 A Shimadzu liquid chromatograph (LC-20AT, Shimadzu, Columbia, MD) equipped with a tandem mass spectrometer (API 3200 ESI/MS/MS system, Applied Biosystems, Foster City, CA) was employed for the detection and quantification of ATCA. Flow injection analysis (FIA) was performed using a flow rate of 0.1 mL/min, a mobile phase consisting of 0.5% acetic acid in methanol, and an injection volume of 20 μL. An ultrasonic cleaner was employed to assist the elution process.2.2 Preparation of polymethacrylic acid (PMAA) grafted silica cylinders (PMAA/silica cylinders)
Methacrylic acid was grafted onto silica cylinder (cut in 1 cm length from silica capillary tubes), and allowed to polymerize by modifying the procedure outlined by Gao et al.18,19 10 g of silica cylinders were placed in 100 mL of aqueous solution containing 5% methane sulfonic acid and activated by stirring (100 rpm) at 102 °C for 4 h. The silica cylinders were rinsed with deionized water, then sonicated for 5 min in deionized water, and dried in an oven at 100 °C. Fifteen mL of the coupling agent MPS and 1.44 g of activated silica cylinder were added to 200 mL of ethanol and water (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). This solution was degassed for 20 min, then purged with nitrogen for 30 min, and finally allowed to react for 24 h at 50 °C while stirring (60 rpm). The silica cylinders were removed for thorough cleaning with methanol, and dried in an oven at 100 °C. The full 1.44 g of MPS/silica cylinders were suspended in a solution containing 20 g of methacrylic acid, 400 mL of deionized water, and 0.12 g ammonium persulfate. This solution was degassed for 20 min and the polymerization was carried out under a nitrogen atmosphere for 7 h at 70 °C while stirring (70 rpm). The silica cylinders were removed and rinsed with methanol, and finally the PMAA/silica cylinders were dried in an oven at 100 °C.
:1). This solution was degassed for 20 min, then purged with nitrogen for 30 min, and finally allowed to react for 24 h at 50 °C while stirring (60 rpm). The silica cylinders were removed for thorough cleaning with methanol, and dried in an oven at 100 °C. The full 1.44 g of MPS/silica cylinders were suspended in a solution containing 20 g of methacrylic acid, 400 mL of deionized water, and 0.12 g ammonium persulfate. This solution was degassed for 20 min and the polymerization was carried out under a nitrogen atmosphere for 7 h at 70 °C while stirring (70 rpm). The silica cylinders were removed and rinsed with methanol, and finally the PMAA/silica cylinders were dried in an oven at 100 °C.
2.3 Preparation of ATCA molecular imprinted material (MIP-PMAA/silica cylinders)
Eight of the PMAA/silica cylinders were suspended in 50 mL of aqueous solution containing 1% acetic acid and 1173 μg/mL ATCA. This was allowed to react at room temperature for 6 h on a shaker, and then dried in an oven at 100 °C. The ATCA-absorbed PMAA/silica cylinders were suspended in 50 mL of ethanol and water (v/v = 1:1) containing 1% acetic acid, 603 μg/mL ATCA, and 0.1 g of EGDE. Note that epoxy coupling in a basic media is a more common procedure. However, such coupling would also occur under acidic conditions.22,23 This was allowed to react in an oven at 50 °C for 8 h. The silica cylinders were rinsed with methanol. Finally the MIP-PMAA/silica cylinders were dried in an oven at 100 °C. The template was removed from the cylinders by washing with 0.5% acetic acid in methanol.
2.4 Evaluation of molecular imprinting by consecutive desorptions
A single MIP-PMAA/silica cylinder was placed in a sample vial containing 200 μL of 0.5% acetic acid in methanol and sonicated for 5 min. To desorb ATCA from the MIP-PMAA/silica cylinder, this was repeated five times. The ATCA concentrations in these desorption solutions were then measured by FIA-ESI/MS/MS immediately.2.5 Construction of stir bars for MISBSE
The MISBSE stir bar was constructed by placing a MIP-PMAA/silica cylinder (1 cm long) over the threaded section of a hex screw, before securing it in place using the matching bolt. The effect of stirring rate on the MISBSE efficiency was assessed next. The stir bar was used to stir each 10 mL of ATCA solution (400 ng/mL) at 100 rpm to 1000 rpm, for 30 min. After MISBSE, 200 μL of 0.5% acetic acid in methanol was used to desorb ATCA from the stir bar. Regeneration of MISBSE stir bar was confirmed when no ATCA was detected in the final desorption solution.2.6 Optimization of MISBSE time
The time needed for the MISBSE to reach equilibrium was assessed. The stir bar was used in 400 ng/mL ATCA standard solutions for 10, 20, 30, 40, 50, and 60 min while stirring at 700 rpm. Note that 700 rpm was a good stirring rate as determined from earlier experiments. Afterwards, ATCA was desorbed from the stir bar with 200 μL of 0.5% acetic acid in methanol for FIA-ESI/MS/MS.2.7 Selectivity of imprinted and non-imprinted stir bars
A non-imprinted polymer (NIP)-PMMA/silica cylinder was prepared by using the same polymerization procedure for the preparation of MIP-PMMA/silica cylinder but in the absence of the template molecule.24,25 This means both MIP and NIP stir bars should have the same polymer compositions but possibly different polymer morphologies.26 In order to demonstrate that the new surface imprinting technique could offer imprinting effect (i.e. shape, size, and functionality) for selective MISBSE in the rebinding of ACTA, the NIP-PMAA/silica cylinder was used for non-imprinted stir bar sorption extraction (NISBSE).42 μL aliquots of 95 μg/mL ATCA stock solution (containing 0.5% acetic acid in methanol) were placed in three 20 mL vials and dried down. 10 mL aliquots of 1% acetic acid in urine, obtained from a nonsmoker, were added to the vials. Three other vials were only filled with 10 mL of 1% aqueous acetic acid in urine. ATCA was extracted from these samples by MISBSE and NISBSE for 30 min at 700 rpm. Before the final desorption of ATCA, a washing step was performed to remove interferences from the stir bars. Different percentages of acetic acid in methanol were tested.
2.8 Optimal source-dependent and compound-dependent parameters of ESI/MS/MS
In order to detect ATCA by ESI/MS/MS, source-dependent and compound-dependent parameters of ESI/MS/MS must be first optimized. A 5 μg/mL ATCA standard solution was infused to the MS/MS at 10 μL/min in order to exam certain compound-dependent parameters for multiple reaction monitoring (MRM) transitions. These compound-dependent parameters include declustering potential (DP), entrance potential (EP), collision cell entrance position (CEP), collision energy (CE), and collision cell exit position (CXP). After optimization of compound-dependent parameters, source-dependent parameters, such as curtain gas (CUR), ionspray voltage (IS), nebulizer gas pressure (Gas 1), turbo gas pressure (Gas 2), collision gas pressure (CAD) and ion source temperature can then be optimized by FIA of ATCA standard solution. Note that these parameters are common parameters that need to be optimized for any ESI/MS/MS instrument before analysis. When these parameters are determined, another person who does the analysis can follow the optimal parameters for ATCA analysis without re-evaluating or re-tuning. The optimal conditions for the detection of ATCA by ESI/MS/MS are described in Section 3.1.3. Results and discussion
3.1 ESI/MS/MS conditions and flow rate selection
When ATCA standard solution was infused to the electrospray ionization (ESI) source under optimal ESI conditions, the production of ATCA molecular ion at m/z 147, which corresponded to [M + H]+, could be detected. The API 3200 ESI/MS/MS system was also used under the MRM scan mode in order to find an optimal condition for the detection of product ions from the molecular ion. Product ion at m/z 101 was strong after the optimization of compound-dependent parameters of MRM, hence the transition ion m/z 147 → 101 was selected for identification and quantification of ATCA. Optimized compound-dependent and source-dependent parameters of ESI/MS/MS for the production of ATCA transition ion, m/z 147 → 101, were DP: 26.00 volts, EP: 9.50 volts, CEP: 14.00 volts, CE 17.00 volts, CXP 4.00 volts, CUR: 50.00 psi, IS: 5500.00 volts, Gas 1: 70.00 psi, Gas 2: 20.00 psi, CAD: 6.00 psi, and ion source temperature: 450 °C.After optimization of ATCA transition ion for the MRM detection, in order to optimize the sensitivity of FIA, mobile phase flow rates of 0.1 mL/min and 0.5 mL/min were tested. Serial dilution was performed, resulting in standard solutions of ATCA ranging from 0.004 μg/mL to 0.125 μg/mL which were used to prepare standard calibration curves. From the calibration data shown in Fig. 1, a lower flow rate produced a sharper slope of calibration curve (y = 431542x2 + 2E + 06x + 43320, R2 = 0.999) compared to the calibration curve produced by the higher flow rate (y = 749419x + 8000, R2 = 0.998). Hence, the flow rate of 0.1 mL/min was selected for all analysis.
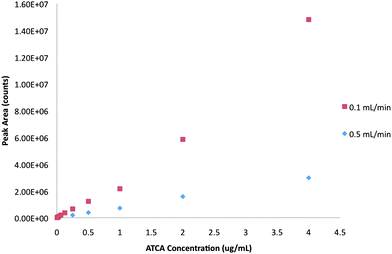 | ||
| Fig. 1 The comparison of the calibration curves of ATCA obtained from FIA-ESI/MS/MS at the flow rate of 0.1 mL/min and 0.5 mL/min. | ||
3.2 Removal of ATCA from MIP-PMAA/silica cylinder by consecutive desorptions
Removal of ATCA template was first evaluated by consecutive desorptions with 200 μL of 0.5% acetic acid in methanol. Any ATCA desorbed from the MIP-PMMA/silica cylinder was measured by FIA-ESI/MS/MS. The peak area of the m/z 147 → 101 ion was used for the identification and quantification of ATCA. Fig. 2 shows a decreasing trend of ATCA as the number of desorptions was increased. This result not only demonstrates a successful imprinting of ATCA on to the silica cylinder, but also suggests the effectiveness of using 0.5% acetic acid in methanol for ATCA desorption. Note that bare silica cylinders were also used for SBSE of ATCA. The results showed no pre-concentration of ATCA onto the bare silica cylinder. Hence, the presence of unreacted silanol groups on the silica surface would not interfere with the MISBSE.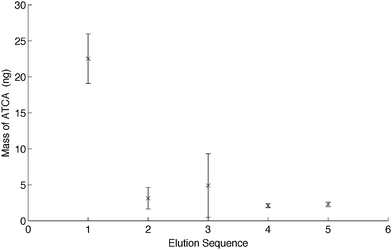 | ||
| Fig. 2 The consecutive desorptions performed on MIP stir bars right after the synthesis of MIP-PMAA on the silica cylinders (n = 3). | ||
Further analysis of the data indicated that approximately 62% of the ATCA present in each MIP-PMAA/silica cylinder was desorbed in the first 200 μL elution, which can be considered as a good recovery for the analysis. More than 80% of ATCA could be desorbed from the MIP by two elutions. All of the ATCA could be completely desorbed when 1.2 mL was used. Note that this quantitative desorption of ATCA was compared to the binding capacity of one MISBSE. The total amount of ATCA desorbed from five consecutive desorptions was determined to be 35 ± 3 ng (n = 3). This number can be translated into a capacity of 35 ng of ATCA per MIP-PMAA/silica cylinder. Both ATCA and MAA have the carboxyl moiety, with a pKa of about 4.3. It was not difficult to see that addition of acetic acid in methanol reduced the interaction of ATCA with MAA during the desorption step.
Since the ESI/MS/MS was very sensitive for ATCA detection with a linear dynamic range of 5 to 100 ng/mL, the present MISBSE-ESI/MS/MS method is more than adequate for the analysis of endogenous levels of ATCA. This sensitivity was compared to a fluorometric determination of ATCA developed by Lundquist et al.,10 where a linear dynamic range of 0.3 to 25 μM (43.8 to 3650 pg/μL) was reported. No correlation coefficient was attempted for FIA-ESI-MS/MS versus the Lundquist method.
3.3 The effect of stirring rate and extraction time on MISBSE
As shown in Fig. 3, an increase of ATCA was found at 700 rpm for MISBSE. When the stir bar rotation speed was greater than 700 rpm, there was a decrease of ATCA. The rapid decrease of ATCA at rotation speeds above 700 rpm might be the high degree of disturbance between the liquid phase and the solid phase, which caused inadequate contact between the molecular binding sites and the target analyte. It might also be due to polymer deformation. The decrease in retention was reversible. This MISBSE was reused and continued to exhibit good extractive capabilities in subsequent experiments. The optimal MISBSE stirring rate was slower than the stirring rate of 1000 rpm used in the polydimethylsiloxane (PDMS) stir bar extraction method by other researchers.27 This variation was probably due to a difference in the overall surface area of the stir bar they used.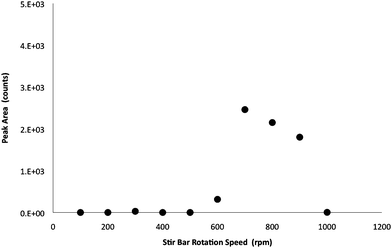 | ||
| Fig. 3 Assessment of the effects of stirring rates on the MISBSE for ATCA. (Extraction time: 45 min.) | ||
In the optimization of extraction time for MISBSE, 30 min seemed to be an adequate amount of time for extraction. As shown in Fig. 4, an increase of extraction of ATCA was found between 20 min and 30 min. Maximum extraction was observed when extraction times went beyond 30 min. The MISBSE extraction time was shorter than the extraction time reported in another MIP stir bar extraction method by Zhu et al.17 It suggested that the covalent grafting of polymer on the surface of silica cylinder created surface-distributed molecular binding sites. These binding sites allowed fast kinetics for the extraction of analyte molecules in the sample solution.
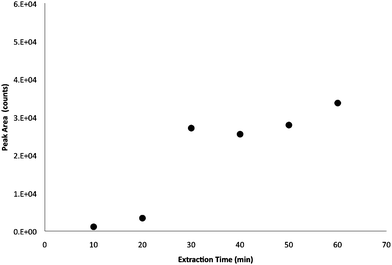 | ||
| Fig. 4 The assessment of the effects of extraction time on the MISBSE for ATCA. (Stir rate: 700 rpm.) | ||
3.4 Selectivity of MISBSE
To evaluate cross-selectivity/imprinting effect, the template and its analogues are often used in competitive rebinding processes.20 In this work, differential pulsed elution (DPE) was adopted, since DPE would easily determine the relative strength among different elution solutions.28–30 As shown in Fig. 5, the MISBSE offered a higher recovery of ATCA from the urine sample. It demonstrated a better selectivity for ATCA in urine samples by MISBSE when compared to NISBSE. Also, the first desorption, using 0.5% acetic acid in methanol, of MISBSE was able to desorb only 20% of the ATCA. In contrast, when no interferences (due to the urine matrix) were present, 62% ATCA could be desorbed by the desorption solution, as shown in Fig. 2. This discrepancy suggested that interferences non-specifically adsorbed on the MISBSE stir bar surface and effectively blocked the accessibility of desorption solution to the molecular imprint binding sites. After removal of interferences from the MISBSE stir bar surface by the washing step, ATCA was successfully eluted by the second desorption (which was also the final desorption). This observation was also supported by the comparison of a larger amount of ATCA desorbed from NISBSE in a spiked urine sample compared to the MISBSE in Fig. 5. Note that the urine sample tested in this experiment was not corrected for creatinine or any other urinary marker. Even though the NISBE seemed to retain as much ATCA as the MISBSE in the spiked urine sample, a better selectivity of MISBSE over NISBSE was demonstrated by differential elution during the washing step. Furthermore, the MISBSE could extract more ATCA from unspiked urine compared to that from NISBSE when combining extracted ATCA from both washing step and final elutions. Note that washing solution and desorption solution had the same acetic acid concentration. The data shown in Fig. 5 was obtained from a single MISBSE and NISBSE. These stir bars were not reused for this experiment. Table 1 displays promising preliminary results suggesting that, given identical experimental conditions, the ATCA recovery for MISBSEs is approximately twice that for NISBSEs. Experiments incorporating internal standards and higher levels of replication are ongoing. Due to variations of recovery, for quantitative analysis of ATCA in real urine samples (non-spiked urine), further research is being performed as a part of our studies to identify a proper internal standard.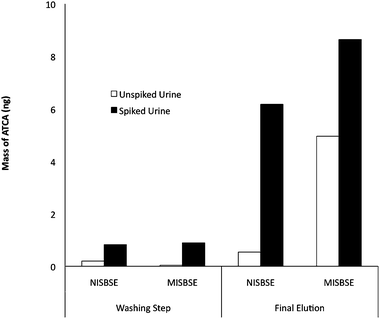 | ||
| Fig. 5 Extraction of ATCA between a NISBSE (non-imprinted PMAA/silica stir bar) and a MISBSE (MIP-PMAA/silica stir bar). Selectivity of MISBSE for ATCA was demonstrated by the intermediate washing step before final desorption. | ||
| Stir Bar | Peak Area (counts) | Average of Peak Area | Recovered ATCA mass (ng) | Comparison of Recovered ATCA Mass by MISBSE/NISBSE |
|---|---|---|---|---|
| MISBSE 1 | 8.77E + 03 | |||
| MISBSE 2 | 6.93E + 03 | 7.85E + 03 | 35.3 | |
| NISBSE 1 | 1.21E + 03 | |||
| NISBSE 2 | 9.88E + 02 | 1.10E + 03 | 19.3 | |
| 1.82 | ||||
These observations suggested the MISBSE offered higher affinity towards ATCA, and clearly indicated that the imprinting effect (due to shape, size and functionality) facilitated the MISBSE rebinding process with ACTA, rather than the simple hydrophobic affinity interaction in NISBSE where shape and size effects are negligible.
4. Conclusions
A MISBSE stir bar was successfully prepared by a grafting imprinting technique. The performance of the MISBSE was demonstrably adequate for the extraction of ATCA at the pg/μL concentration level. Although moderate selectivity was achieved, this work represents a successful preparation of selective solid phase extraction materials by molecular imprinting, particularly on silica surface for SBSE. The combination of MIP and SBSE is of great interest for the development of modern analytical techniques in forensic toxicology, pharmaceutical analysis and biomedical diagnostics. In addition, with the advanced instrumentation of electrospray ionization tandem mass spectrometry (ESI/MS/MS), ATCA could be detected without the use of any derivatization process. This new strategy of MISBSE-ESI/MS/MS improved the selectivity and sensitivity for the detection of trace ACTA in urine samples. Due to the selectivity of MS/MS, other urinary acids absorbed or retained by MISBSE did not interfere the detection of ATCA. However, the effect of protein binding and optimal urinary pH for extraction will be part of future investigation efforts (to achieve better than 62% recovery). Nonetheless, the column-less method showed promise in further advancement of cyanide exposure research. Future work in our lab will focus on application of this MISBSE technique to determine endogenous ATCA, and hence a cutoff limit for smokers, in order to identify a point at which cyanide exposure warrants both medical attention and forensic investigation.Acknowledgements
The authors are thankful to Dr Brian Logue (Department of Chemistry and Biochemistry, Shepard Hall, Brooking, SD, USA) for providing ATCA for these studies. This study was supported by the Sam Houston State University, Huntsville, TX, and NIH: NIAID/USAMRICD Interagency Agreements (Y1-A1-6176-01/02 and A120-B.P2006/7-01), and the U.S. Army Medical Research Institute Of Chemical Defense (Dr Gary Rockwood) under the auspices of the U.S. Army Research Office Scientific Services Program administered by Battelle (Delivery Order 0878, Contract No. DAAD19-02-D-0001, TCN 08284). The technical assistance of Kelsie Simons is also greatly acknowledged.References
- S. Baskin and T. Brewer, in Textbook of Military Medicine, Office of the Surgeon General, ed. F. Sidell, E. Takafuji and D. Franz, Falls Church, VA, 1997, ch. 10, pp. 271–286 Search PubMed.
- B. A. Logue, N. P. Kirschten, I. Petrikovics, M. A. Moser, G. A. Rockwood and S. I. Baskin, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2005, 819, 237–244 CrossRef CAS.
- S. I. Baskin, I. Petrikovics, G. E. Platoff, G. A. Rockwood and B. A. Logue, Toxicol. Mech. Methods, 2006, 16, 339–345 CrossRef CAS.
- F. Moriya and Y. Hashimoto, J. Forensic Sci., 2001, 46, 1421–1425.
- S. I. Baskin, I. Petrikovics, J. S. Kurche, J. D. Nicholson, B. A. Logue, B. J. Maliner and G. A. Rockwood, Pharmacological Perspectives of Toxic Chemicals and Their Antidotes, 2004 Search PubMed.
- B. Ballantyne, Clin. Toxicol., 1977, 11, 173–193 Search PubMed.
- S. I. Baskin and G. E. Isom, in Comprehensive Toxicology, ed. I. G. Sipes, C. A. McQueen and A. J. Gandolfi, Elsevier Science, New York, 1997, 477–488 Search PubMed.
- W. A. Himwich and J. P. Saunders, Am. J. Physiol., 1948, 153, 348–354 Search PubMed.
- B. A. Logue, B. J. Pieper, I. Petrikovics, M. A. Moser, N. P. Kirchten, S. I. Baskin and G. A. Rockwood, Medical Defense Bioscience Review (CD-ROM), U.S. Army Medical Research Institute of Chemical Defence. APG, MD, 2004 Search PubMed.
- P. Lundquist, B. Kagedal, L. Nilsson and H. Rosling, Anal. Biochem., 1995, 228, 27–34 CrossRef CAS.
- B. A. Logue, W. K. Maserek, G. A. Rockwood, M. W. Keebaugh and S. I. Baskin, Toxicol. Mech. Methods, 2009, 19, 202–208 CrossRef CAS.
- M. Yan and O. Ramstrom, Molecularly Imprinted Materials: Science and Technology, Marcel Dekker, New York NY, 2005 Search PubMed.
- K. Kim and D. Kim, J. Appl. Polym. Sci., 2005, 96, 200–212 CrossRef CAS.
- K. Haupt, A. G. Mayes and K. Mosbach, Anal. Chem., 1998, 70, 3936–3939 CrossRef CAS.
- M. Ávila, M. Zougagh, R. Á and A. Escarpa, TrAC, Trends Anal. Chem., 2008, 27, 54–65 CrossRef CAS.
- E. Baltussen, P. Sandra, F. David and C. Cramers, Editon edn, 1999, vol. 11, p. 747.
- X. Zhu, J. Cai, J. Yang, Q. Su and Y. Gao, J. Chromatogr., A, 2006, 1131, 37–44 CrossRef CAS.
- B. Gao, R. Wang, H. Jiu and D. Kong, J. Appl. Polym. Sci., 2006, 102, 5808–5817 CrossRef CAS.
- B. Gao, J. Wang, F. An and Q. Liu, Polymer, 2008, 49, 1230–1238 CrossRef CAS.
- M. Husseman, E. E. Malmstrom and M. McNamara, Macromolecules, 1999, 32, 1424–1431 CrossRef.
- H. T. Nagasawa, S. E. Cummings and S. I. Baskin, Org. Prep. Proced. Int., 2004, 36, 178–182 CrossRef CAS.
- F. Heba, M. Mouzali and M. J. M. Abadie, J. Appl. Polym. Sci., 2003, 90, 2834–2839 CrossRef CAS.
- Y. Li, K. Moon and C. P. Wong, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1020–1027 CrossRef CAS.
- X. Jiang, W. Tian, C. Zhao, H. Zhang and M. Liu, Talanta, 2007, 72, 119–125 CrossRef CAS.
- E. Papaioannou, C. Koutsas and M. Liakopoulou-Kyriakides, Amino Acids, 2009, 36, 563–569 CrossRef CAS.
- K. Haupt and K. Mosbach, Chem. Rev., 2000, 100, 2495–2504 CrossRef CAS.
- M. Kawaguchi, R. Ito, H. Honda, N. Endo, N. Okanouchi, K. Saito, Y. Seto and H. Nakazawa, J. Chromatogr., A, 2008, 1206, 196–199 CrossRef.
- W. Mullett and E. Lai, Anal. Chem., 1998, 70, 3636–3641 CrossRef CAS.
- W. Mullett, E. Lai and B. Sellergren, Anal. Commun., 1999, 36, 217–220 RSC.
- J. C. C. Yu, A. Hrdina, C. Mancini and E. P. C. Lai, J. Nanosci. Nanotechnol., 2007, 7, 3095–3103 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |