DOI:
10.1039/B9AY00276F
(Paper)
Anal. Methods, 2010,
2, 525-531
HPTLC method development and validation: Quantification of paliperidone in formulations and in vitro release study
Received
29th November 2009
, Accepted 1st February 2010
First published on
12th February 2010
Abstract
A new, economic, environmentally friendly and rapid high-performance thin-layer chromatographic (HPTLC) method was developed and validated for quantitative determination of paliperidone. The HPTLC separation was achieved on an aluminium-backed layer of silica gel 60F254 using methanol–ethyl acetate (8.0 + 2.0 v/v) as mobile phase. Quantitation was achieved by densitometric analysis at 284 nm over the concentration range of 100–600 ng mL−1. The method was found to give compact spot for the drug (Rf 0.54 ± 0.011). The linear regression analysis data for the calibration plots showed good linear relationship with r2 = 0.9997. The method was validated for precision, recovery, repeatability, and robustness as per the International Conference on Harmonization guidelines. The minimum detectable amount was found to be 15.11 ng/spot, whereas the limit of quantitation was found to be 45.79 ng/spot. Statistical analysis of the data showed that the method is precise, accurate, reproducible, sensitive and selective for the analysis of paliperidone. The method was successfully employed for the estimation of paliperidone as a bulk drug, equilibrium solubility study, commercially available tablet formulation, mucoadhesive microemulsion formulations and solution (developed in-house).
Introduction
Paliperidone (PPD), (±)-3-{2-[4-(6-Fluoro-1,2-benzisoxazol-3-yl)piperidino]ethyl}-6,7,8,9-tetrahydro-9-hydroxy-2-methyl-4H-pyrido[1,2-a]pyrimidin-4-one is a primary active metabolite of older antipsychotic drug resperidone. PPD is indicated for acute and maintenance treatment of schizophrenia.1–5
PPD is marketed by Janssen under the brand name of Invenga™ in 3 mg, 6 mg and 9 mg doses.1 These dosage forms exhibit low bioavailability due to extensive first-pass metabolism, and the nontargeted delivery results in numerous side effects. As the target site of the PPD is brain, thereby a strategy is desirable that not only improves the bioavailability by preventing first pass metabolism but also provides targeting to the receptor site and bypasses the blood–brain barrier so as to achieve desired drug concentration at the site of action, hence preventing the availability of drug at nontargeting sites and reducing the side effects. Earlier studies6,7 have demonstrated direct transport of drugs to brain, circumventing the brain barriers following intranasal (i.n.) administration that provides a unique feature and better option for targeting drugs to brain.8,9 Microemulsion (ME) by virtue of their lipophilic nature and small globule size10–12 is widely explored as a delivery system to enhance uptake across nasal mucosa and the addition of mucoadhesive agents such as polyelectrolyte polymer helps in the retention of the formulation on the nasal mucosa.14
The review of literature revealed that methods of analysis for PPD in pharmaceutical formulation has not been reported before. During preliminary studies, drug content in the formulations was determined by UV–Visible spectrometric method and it was found that the excipients present in the mucoadhesive microemulsion (MME) formulation interfered in the estimation of drug. Further, HPLC based separation methods may not be suitable for the determination of drug from lipid-based delivery systems such as MME formulations. These formulations contain various lipophilic excipients that are not soluble in commonly used organic solvents used in HPLC methods. Further, extraction of drug from such lipophilic excipients may not be achieved easily, and such excipients may get adsorbed on the stationary phase. Hence, analysis of PPD, particularly from lipid based delivery systems, would be difficult with respect to identification of suitable solvents and stationary phase.
In view of this, high-performance thin layer chromatography (HPTLC) based methods could be considered as a good alternative as they are being explored as an important tool in routine drug analysis. A major advantage of HPTLC is its ability to analyze several samples simultaneously using a small quantity of mobile phase; this reduces the time and cost of analysis.13–15 In addition, it minimizes exposure risks and significantly reduces disposal problems of toxic organic effluents, thereby reducing possibilities of environment pollution. HPTLC also facilitates repeated detection of chromatogram with the same or different parameters. Furthermore, in the case of HPTLC, there are no restrictions on the choice of solvents and mobile phases; drug and lipophilic excipients can be dissolved in a suitable solvent that would evaporate during spotting on HPTLC plate leaving behind analyte as a thin band. Therefore, for such methods, extraction procedure is not required always and could be developed for analyzing drug without any interference from excipients.13–15 To our present knowledge, HPTLC method of analysis has not been explored for PPD before. Therefore it was felt necessary to develop a HPTLC method for determination and quantitative estimation of PPD. In view of this, the present study describes the development of environment friendly, simple, rapid, economic, and validated HPTLC method for routine estimation of PPD from bulk and pharmaceutical dosage forms such as tablets and MME formulations and solution developed in-house.
Experimental
Apparatus
The HPTLC system (Camag, Muttenz, Switzerland) consisted of Limomat V autosprayer connected to a nitrogen cylinder, a twin trough chamber (10 × 10 cm), a derivatization chamber, and a plate heater. Pre-coated silica gel 60 F254 TLC plates (10 × 10 cm, layer thickness 0.2 mm (E. Merck KGaA, Darmstadt, Germany)) were used as stationary phase. TLC plates were pre-washed twice with 10 mL of methanol and activated at 80 °C for 5 min prior to sample application. Densitometric analysis was carried out using a TLC scanner III with winCATS software.
Reagents and materials
PPD pure powder was obtained as gratis sample from Torrent Pharmaceutical (Ahmedabad–India) with 99.9% purity. Tablet formulation, Pali XR 6 (Intas Pharmaceuticals, India) was obtained commercially with the labelled amounts of 6.0 mg of PPD. Labrafac CC (Caprylic/Capric Triglycerides, C8–C10 fatty acids), Labrasol (caprylocaproyl macrogol-8-glyceride), Plurol Oleique (Polyglyceryl 6-dioleate) (Gattefosse Saint-Priest, France) was procured as gratis sample from Gattefosse Asia Ltd. (Mumbai, India). Polycarbophil (AA-1, pharmagrade, molecular weight approximately 3.5 million) was procured as gratis sample from Lubrizol Advance Material India Pvt Ltd. (Mumbai, India). Potassium dihydrogen phosphate, methanol, propylene glycol were purchased from SDfine Chemicals (Ahmedabad, India). Ethanol was purchased from Baroda Chemical Ind. Ltd (Dabhoi, India). Double distilled water was used throughout the study. All other chemicals and solvents were of analytical reagent grade and used as received without further purification.
HPTLC method and chromatographic conditions
Sample application.
The standard and formulation samples of PPD were spotted on pre-coated TLC plates in the form of narrow bands of lengths 6 mm, with 10 mm from the bottom and left margin and with 9 mm distance between two bands. Samples were applied under continuous drying stream of nitrogen gas at constant application rate of 150 nL s−1.
Mobile phase and migration.
Plates were developed using mobile phase consisting of methanol–ethyl acetate (8.0 + 2.0 v/v). Linear ascending development was carried out in 10 cm × 10 cm twin trough glass chamber equilibrated with mobile phase. The optimized chamber saturation time for mobile phase was 20 min at 25 ± 2 °C. Ten millilitres of the mobile phase (5 mL in trough containing the plate and 5 mL in other trough) was used for each development and allowed to migrate a distance of 70 mm, which required 10 min. After development, the TLC plates were dried completely.
Densitometric analysis and quantitation procedure.
Densitometric scanning was performed on Camag TLC scanner III in absorbance mode and operated by winCATS planar chromatography version 1.3.4. The source of radiation utilized was deuterium lamp. The spots were analyzed at a wavelength of 284 nm. The slit dimensions used in the analysis were length and width of 5 mm and 0.45 mm, respectively, with a scanning rate of 20 mm s−1. These are selected as recommended by the CAMAG TLC Scanner III manual. It covers 70–90% of the application band length, which in the present case is 6 mm. The monochromator bandwidth was set at 20 nm. Concentrations of compound chromatographed were determined from the intensity of diffusely reflected light and evaluated as peak areas against concentrations using linear regression equation.
Preparation of PPD standard stock solution.
Stock solution was prepared by weighing PPD (10 mg). Weighed powder was accurately transferred to a volumetric flask of 100 mL and dissolved in and diluted to the mark with methanol to obtain a standard stock solution of PPD (100 μg mL−1).
Method validation.
Validation of the developed HPTLC method was carried out as per the International Conference on Harmonization (ICH) guidelines Q2 (R1) for specificity, sensitivity, accuracy, precision, repeatability, and robustness.16
Specificity.
The specificity of the developed method was established analyzing the sample solutions containing PPD from MME formulations and marketed tablets in relation to interferences from formulation ingredients. The spot for PPD in the sample was confirmed by comparing retardation factor (Rf) values of the spot with that of the standard.
Sensitivity.
Sensitivity of the method was determined with respect to limit of detection (LOD) and limit of quantification (LOQ). Noise was determined by scanning blank spot (methanol) six times. Series of concentrations of drug solutions (10–600 ng/spot) were applied on plate and analyzed to determine LOD and LOQ. LOD was calculated as 3 times the noise level, and LOQ was calculated as 10 times the noise level. LOD and LOQ were experimentally verified by diluting the known concentrations of PPD until the average responses were approximately 3–10 times the standard deviation (SD) of the responses for six replicate determinations.
Linearity and calibration curve.
Linearity of the method was evaluated by constructing calibration curves at six concentration levels. Aliquots of standard working solution of PPD were applied to the plate to obtain concentration in the range of 100, 200, 300, 400, 500, and 600 ng/spot. The calibration curves were developed by plotting peak area vs. concentrations (n = 6) with the help of the win-CATS software.
Accuracy.
Accuracy of the method was evaluated by carrying the recovery study at three levels. Recovery experiments were performed by adding three different amounts of standard drug, i.e., 80, 100, and 120% of the drug, to the preanalyzed MME formulations, solution and conventional tablets, and the resultant was reanalyzed six times.
Precision.
Precision was evaluated in terms of intra-day and inter-day precisions. Intra-day precision was determined by analyzing sample solutions of PPD from MME formulations at three levels covering low, medium, and higher concentrations of calibration curve for five times on the same day. Inter-day precision was determined by analyzing sample solutions of PPD at three levels covering low, medium, and higher concentrations over a period of seven days (n = 5). The peak areas obtained were used to calculate mean and %RSD (relative SD) values.
Repeatability (system precision).
Repeatability of measurement of peak area was determined by analyzing different amount of PPD samples covering low, medium, and higher ranges of the calibration curve seven times without changing the position of plate. Repeatability of sample application was assessed by spotting PPD samples covering similar range of calibration curve seven times and analyzing them once.
Robustness.
By introducing small changes in mobile phase composition, its volume, chamber saturation time, and slight change in the solvent migration distance, the effects on the results were examined. Robustness of the method was determined in triplicate at a concentration level of 300 ng/spot and the mean and %RSD of peak area was calculated.
Application of developed method
Determination of equilibrium solubility.
Solubility of PPD in various excipients was determined by shake flask method. An excess of PPD was added to 1 g of each of the excipient and vortexed to facilitate the mixing. Mixtures were shaken for 48 h in a reciprocating water bath shaker maintained at room temperature. After 48 h, each tube was centrifuged at 600g for 10 min, and the insoluble drug was discarded by filtration using a 0.45 μm membrane filter. The filtrate was suitably diluted with methanol and concentration of drug was quantified by a developed HPTLC method.
PPD formulations.
Two MME formulations of PPD were developed in-house, one by mixing labrasol and plurol oleique in a weight ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, with 12% (wt/wt) Labrafac CC and 0.50% (wt/wt) polycarbophil AA-1 (formulation F1: for intranasal delivery of PPD), and another by mixing labrasol and plurol oleique in a weight ratio of 4:1, with 12% (wt/wt) labrafac CC (formulation F2: for intranasal delivery of PPD). Both formulations on dilution with water yield ME with mean particle size less than 27 nm as determined by photon correlation spectroscopy with in-built Zetasizer (Nano ZS, Malvern Instruments, UK) at 633 nm. Helium–neon gas laser having intensity of 4 mW was the light source. PPD solution was prepared by dissolving it in mixture propylene glycol and ethanol weight ratio 7:3 (formulation F3: for both intranasal and intravenous delivery of PPD). Commercially available tablets of PPD, 6 mg were also used in this investigation to verify the suitability of the method for analysis of PPD from conventional dosage forms.
:1, with 12% (wt/wt) Labrafac CC and 0.50% (wt/wt) polycarbophil AA-1 (formulation F1: for intranasal delivery of PPD), and another by mixing labrasol and plurol oleique in a weight ratio of 4:1, with 12% (wt/wt) labrafac CC (formulation F2: for intranasal delivery of PPD). Both formulations on dilution with water yield ME with mean particle size less than 27 nm as determined by photon correlation spectroscopy with in-built Zetasizer (Nano ZS, Malvern Instruments, UK) at 633 nm. Helium–neon gas laser having intensity of 4 mW was the light source. PPD solution was prepared by dissolving it in mixture propylene glycol and ethanol weight ratio 7:3 (formulation F3: for both intranasal and intravenous delivery of PPD). Commercially available tablets of PPD, 6 mg were also used in this investigation to verify the suitability of the method for analysis of PPD from conventional dosage forms.
Analysis of PPD in formulations.
Twenty tablets were weighed and finely powdered. Quantity equivalent to 10 mg of drug was weighed accurately and dissolved in 50 mL methanol. The solution was sonicated for 15 min and then filtered through Whatman filter paper No. 41. The residue was washed thoroughly with methanol. The filtrate and washings were combined and diluted suitably with methanol to obtain a 100 μg mL−1 concentration of PPD. MME formulations containing 10 mg equivalent of PPD were dispersed in 50 mL of methanol and were treated in a similar manner as that of tablets to obtain a stock solution of 100 μg mL−1. A solution containing 10 mg equivalent of PPD was diluted in 50 mL of methanol and was treated in a similar manner as that of tablets to obtain a stock solution of 100 μg mL−1. On TLC plates, 5 μL of these solutions were spotted and analyzed for PPD content using the previously described method. The possibility of interference from other components of the tablet formulation in the analysis was studied. Placebo MME formulations were analyzed similarly to study the potential interference.
In vitro diffusion profile of PPD formulation.
MME formulation of PPD for intranasal delivery (formulation F1 and F2) solution (formulation F3) was evaluated for in vitro release using a Franz diffusion cell with a diameter of 10 mm. The temperature of the receiver chamber containing 15 mL of diffusion media (phosphate buffer, pH 5.0) was controlled at 37 ± 1 °C under continuous stirring with Teflon-coated magnetic bar at a constant rate, in a way that the nasal membrane surface just flushes the diffusion fluid. During study, 2 mL of aliquots were removed at 15, 30, 60, 90, 120, and 240 min and replaced with fresh buffer. Amount of drug released was determined using developed HPTLC method.
Results and discussion
Development of HPTLC method
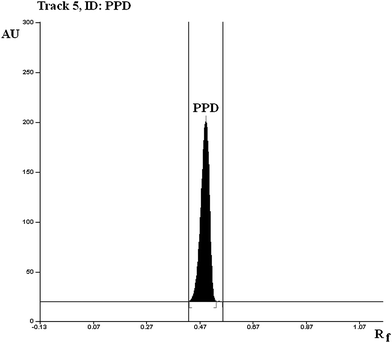
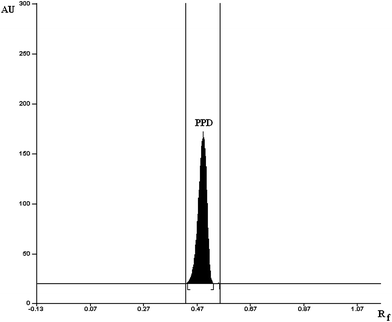
To develop a HPTLC method of analysis for estimation of PPD, selection of mobile phase was carried out on the basis of polarity. A solvent system that would give dense and compact spots with appropriate and significantly different Rf value for PPD was desired. Various solvent systems (such as acetone-methanol, methanol–chloroform, methanol–toluene, methanol–ethyl acetate, toluene–ethyl acetate, hexane–ethyl acetate, hexane–acetone, toluene–acetonitrile, and toluene–acetonitrile–glacial acetic acid) were evaluated in different proportions. Among these, the solvent system comprising of methanol–ethyl acetate (8.0 + 2.0, v/v) gave a peak of PPD at Rf value of 0.54. It was also observed that chamber saturation time and solvent migration distance are crucial in chromatographic separation as chamber saturation time of less than 15 min and solvent migration distances greater than 70 mm resulted diffusion of analyte spot. Therefore, methanol–ethyl acetate solvent system in 8.0 + 2.0 (v/v) proportion with chamber saturation time of 20 min at 25 °C and solvent migration distance of 70 mm was used as mobile phase. These chromatographic conditions produced a well-defined compact spot of PPD on 10 × 10 cm HPTLC silica gel 60F254 aluminium-backed plates with optimum migration at Rf 0.54 ± 0.011 (Fig. 1). It also gave a well-defined compact spot of PPD in analysis of various MME formulations, solution and marketed tablet formulation (Fig. 2).
Method validation
Sensitivity.
Under the experimental conditions employed, the lowest amount of drug that could be detected was found to be 15.11 ng/spot and the lowest amount of drug that could be quantified was found to be 45.79 ng/spot, with RSD <5%.
Specificity.
Specificity is the ability of an analytical method to assess unequivocally the analyte in the presence of sample matrix. PPD was separated from excipients with an Rf of 0.54 ± 0.011. There was no interfering peak at the Rf value of PPD from excipients such as Labrasol, Plurol oleique, Labrafac CC and polycarbophil AA-1 present in MME formulations. In addition, there was no interference from excipients, present in commercial formulation, thereby confirming specificity of method.
Linearity and calibration curve.
Linearity of an analytical method is its ability, within a given range, to obtain test results that are directly, or through a mathematical transformation, proportional to concentration of analyte. The method was found to be linear in a concentration range of 100–600 ng/spot (n = 6), with respect to peak area. Fig. 3 displays three-dimensional overlay of HPTLC densitograms of the calibration spots of PPD at 284 nm. The regression data are reported in Table 1. No significant difference was observed in the slopes of standard curves (ANOVA, p > 0.05).
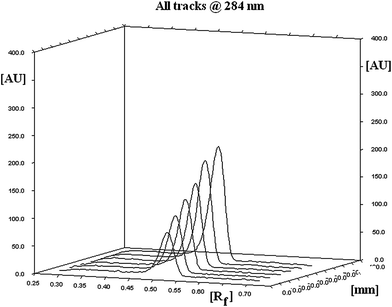 |
| | Fig. 3 Three-dimensional overlay of HPTLC densitograms of calibration spots of Paliperidone. | |
Table 1 Linear regression data for the calibration curves (n = 6)
| Range (ng/spot) |
r2 ± SD |
Slope ± SD |
Intercept ± SD |
| 100–600 |
0.9997 ± 0.00027 |
6.1220 ± 0.076 |
1109.567 ± 28.035 |
Accuracy.
Accuracy of an analytical method is the closeness of test results to true value. It was determined by the application of analytical procedure to recovery studies, where known amount of standard is spiked in preanalyzed samples solutions. The amount of drug recovered in accuracy study was in the range of 98.77–101.39% which indicates that the method is accurate (Table 2).
Table 2 Recovery studies (n = 6)
| Formulation |
Amount of Drug Analyzed/ng |
Amount of Drug Added/ng |
Theoretical Concentration/ng |
Total Amount of Drug Analyzed/ng |
%Recovery ± SD |
| F1 |
200 |
160 |
360 |
356.14 |
98.93 |
| 200 |
200 |
400 |
396.66 |
99.17 |
| 200 |
240 |
440 |
438.36 |
99.63 |
|
|
|
|
%Average recovery ± SD |
99.24 ± 1.05 |
| F2 |
200 |
160 |
360 |
357.16 |
99.21 |
| 200 |
200 |
400 |
398.16 |
99.54 |
| 200 |
240 |
440 |
446.11 |
101.39 |
|
|
|
|
%Average recovery ± SD |
100.05 ± 0.69 |
| F3 |
200 |
160 |
360 |
359.55 |
99.88 |
| 200 |
200 |
400 |
395.09 |
98.77 |
| 200 |
240 |
440 |
440.02 |
100.00 |
|
|
|
|
%Average recovery ± SD |
99.55 ± 1.73 |
| Marketed |
200 |
160 |
360 |
362.69 |
100.75 |
| Tablets |
200 |
200 |
400 |
397.58 |
99.40 |
| 200 |
240 |
440 |
441.23 |
100.28 |
|
|
|
|
%Average recovery ± SD |
100.14 ± 1.19 |
Precision.
The precision of an analytical method expresses the degree of scatter between a series of measurements obtained from multiple sampling of the same homogeneous sample under prescribed conditions. Intra-day precision refers to the use of analytical procedure within a laboratory over a short period of time using the same operator with the same equipment, whereas inter-day precision involves estimation of variations in analysis when a method is used within a laboratory on different days, by different analysts. The %RSD obtained were 1.15–1.45 and 1.04–1.49 for intraday and interday precision study, respectively (Table 3). In all instances, %RSD values were less than 5% confirming the precision of the method.
Table 3 Intra- and Inter-precision studies (n = 5)
| Amount of Drug Spotted/ng |
Amount of Drug Detected (ng, mean ± SD) |
%RSD |
| Intra-day (n = 5) |
| 100 |
99.97 ± 1.09 |
1.15 |
| 300 |
297.96 ± 1.54 |
1.45 |
| 600 |
599.54 ± 1.06 |
1.22 |
| Inter-day (n = 5) |
| 100 |
100.41 ± 0.69 |
1.04 |
| 300 |
297.58 ± 2.67 |
1.19 |
| 600 |
598.77 ± 2.07 |
1.49 |
Repeatability.
Ten-microlitre aliquots of samples containing 100, 300, and 600 ng of PPD were analyzed according to proposed method. In order to control scanner parameters, i.e., repeatability of measurement of peak area, one spot was analyzed without changing position of plate (n = 7). By spotting and analyzing the same amount several times (n = 7), precision of automatic spotting device was evaluated. %RSD was consistently less than 5% (Table 4). The RSD values obtained were in the range of 1.27 to 2.89 which was well below the instrumental specification ensuring repeatability of developed method.
Table 4 Repeatability studies (n = 7)
| Parameters |
Amount of Drug Detected (ng, mean ± SD) |
|
One spot is scanned eight times.
Eight spots scanned once.
|
| Amount of Paliperidone Spotted/ng |
100 |
300 |
600 |
| Measurement of peak areaa |
96.57 ± 5.18 |
295.33 ± 4.95 |
593.69 ± 6.26 |
| %RSD |
1.27 |
2.41 |
2.89 |
| Sample application and derivatization techniqueb |
98.79 ± 2.09 |
301.81 ± 2.70 |
595.66 ± 5.01 |
| %RSD |
1.38 |
2.61 |
2.74 |
Application of developed method
Determination of equilibrium solubility.
PPD has very low water solubility (0.003 g/100 mL), which limits the development of new pharmaceutical formulations aimed to improve its delivery. Therefore, solubility studies were performed to identify suitable oily phases, surfactants, and cosurfactants that are generally employed in commercially available topical, intranasal, parenteral and oral products (Table 6). In spite of being lipophilic in nature, PPD exhibited very low solubility in oily phase such as isopropyl palmitate and miglyols. It was found that only Labrafac CC exhibited good solubility for PPD among the oils studied. The drug exhibited good solubility in Labrasol, Tween 80 (surfactants), Plurol oleique, Transcutol P, Capmul MCM and propylene glycol (cosurfactants). Among the various excipients tried, the oily phases such as Labrafil M 1944 and Labrafac CC exhibited good solubilization potential for PPD. Labrasol and Tween 80 amongst surfactants and plurol oleique, transcutol P and capmul MCM as a cosurfactant were found to solubilize maximum amount of PPD.
Table 6 Solubility of Paliperidone in various excipients and buffers
| Excipients |
Solubilitya |
|
Data expressed as mg g−1, mean ± SD, n = 3.
Data expressed as μg mL−1, mean ± SD, n = 3.
|
|
Oily phases
|
| Labrafil M 1944 (Oleoyl polyoxylglycerides) |
6.21 ± 1.32 |
| Labrafac CC (Caprylic/Capric Triglycerides) |
17.76 ± 3.45 |
| Isopropyl Myristate |
11.05 ± 3.55 |
| Labrafac Lipophile (Medium chain triglycerides) |
0.73 ± 0.31 |
| Labrafac PG (Propylene glycol dicaprylocaprate) |
0.36 ± 0.21 |
| Miglyol 810 (Caprylic/Capric Triglyceride) |
1.04 ± 0.69 |
| Miglyol 812 (Caprylic/Capric Triglyceride) |
1.48 ± 0.91 |
| Miglyol 840 (Propylene Glycol Dicaprylate/Dicaprate) |
1.55 ± 1.01 |
| Lauryl Alcohol |
2.61 ± 1.41 |
| Isostearylic isostearate |
0.70 ± 0.25 |
| Isopropyl Palmitate |
0.95 ± 0.27 |
| Captex 200 (Propylene GlycoDicaprylate/Dicaprate) |
2.47 ± 1.08 |
| Captex 355 (Glycerol Caprylate Caprate) |
0.89 ± 0.77 |
|
Surfactants
|
| Labrasol (Caprylocaproyl Polyoxylglycerides) |
17.44 ± 3.56 |
| Tween (Polysorbate) 80 |
18.38 ± 4.48 |
| Plurol Stearique WL (Polyglyceryl-6-distearate) |
1.00 ± 0.57 |
| Plurol Diisostearique (Polyglyceryl diisostearate) |
0.65 ± 0.36 |
| Cremophor RH 40 (Polyoxyl 40 Hydrogenated Castor Oil) |
9.11 ± 2.35 |
|
Cosurfactants
|
| Plurol Oleique CC (Polyglyceryl oleate) |
13.29 ± 5.34 |
| Plurol Oleique 5203 (Polyglyceryl 6-dioleate) |
1.20 ± 0.34 |
| Lauroglycol 90 (Propylene glycol monolaurate) |
0.96 ± 0.23 |
| Capryol 90 (Propylene glycol monocaprylate) |
0.76 ± 0.61 |
| Transcutol P (Diethylene glycol monoethyl ether) |
49.06 ± 5.33 |
| Capmul MCM (Glyceryl Mono- & dicaprate) |
18.25 ± 3.12 |
| Propylene glycol |
17.49 ± 4.23 |
|
Aqueous Phases
|
| Phosphate buffer pH 5.0 |
31.07 ± 2.65 |
| Phosphate buffer pH 6.0 |
11.26 ± 3.77 |
| Phosphate buffer pH 6.5 |
12.14 ± 3.59 |
Determination of pH-solubility profile.
An understanding of pH-solubility profile of a drug candidate is regarded as one of the most important aspects of preformulation testing for poorly soluble compounds and has been found to be useful for topical, intranasal, oral and parenteral formulation development. For example, nasal secretion exhibits a pH in the range of 4.5–6.5, and the solubility profile of the drug can help in predicting the absorption behaviour of that drug molecule in the nasal mucosa. The study indicated that PPD has adequate aqueous solubility and it depends on pH of the medium (Table 6).
Analysis of PPD in formulations.
A single spot at Rf 0.54 was observed in the chromatogram of PPD. No interference from the excipients present in the marketed tablet formulation was observed. Analysis of PPD tablets showed a drug content of 6.02 ± 0.14 mg. The applicability of the method was verified by the determination of PPD in two MME formulations and solution (developed in-house), and no interference from the excipients matrix was observed. The PPD content of the developed and the marketed formulations was found to be within the limits (±5% of the theoretical value) and are mentioned in Table 7. The low %RSD value indicated the suitability of this method for routine analysis of PPD in various formulations.
Table 7 Content of Paliperidone in various formulations
| Formulation |
Label Claim/mg |
Amount Found (mg, mean ± SD) |
%RSD |
| F1 |
5 |
5.08 ± 0.32 |
1.44 |
| F2 |
5 |
4.65 ± 0.86 |
1.03 |
| F3 |
5 |
5.39 ± 0.54 |
1.52 |
| Tablets |
6 |
6.02 ± 0.14 |
1.08 |
In vitro diffusion profile of PPD formulation.
In vitro diffusion profile of PPD formulation F1, F2 and F3 are presented in Fig. 4. Formulation F3 was found to exhibit a release of 50% drug within 30 min in diffusion media. It was also evident that release of PPD from MME was consistent with the results of pH-solubility profile study.
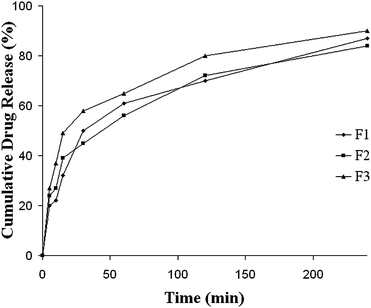 |
| | Fig. 4 Diffusion profiles of Paliperidone from various formulations in buffer pH 5.0, data expressed as mean ± SD, n = 3. | |
Conclusion
A new HPTLC method has been developed for the identification and quantification of PPD. Low cost of ingredients, faster speed, and satisfactory precision and accuracy are the main features of this method. Method was successfully validated as per ICH guidelines and statistical analysis proves that method is sensitive, specific, and repeatable. It can be conveniently employed for routine quality control analysis of PPD as bulk drug in marketed tablets, MME formulations without any interference from excipients. The method was also applied for the estimation of equilibrium solubility of PPD in various excipients and diffusion studies.
Acknowledgements
Authors are thankful to Torrent Pharmaceutical Ltd for the gift sample of PPD pure powder, Sophisticated Instrumentation Center for Applied Research and Testing (SICART) (Vallabh Vidyanagar, India) for providing facilities for carrying out analytical work, Gattefosse (Saint-Priest, France), Colorcon (Asia) Pvt. Ltd. (Mumbai, India), Abitec Corporation (Janesville, USA), BASF (Mumbai, India), Sasol (Witten, Germany), Lubrizol Advance Material India Pvt. Ltd. (Mumbai, India), Noveon (Cleveland, USA) for providing gratis samples of excipients, Mr. Dimal A. Shah, Assit. Prof., Dept. of Pharmaceutical Chemistry, Indukaka Ipcowala College of Pharmacy, for providing technical guidance and for checking the manuscript.
References
- Janssen-Cilag Ltd. Summary of Product Characteristics - Invega® 3 mg, 6 mg, 9 mg, 12 mg prolonged release tablets (Paliperidone) 28/01/2009 (last accessed 08/08/2009).
- M. N. Samtani, A. Vermeulen and K. Stuyckens, Clin. Pharmacokinet., 2009, 48, 585–600 CrossRef CAS.
- C. M. Canuso, C. A. Bossie, I. Turkoz and L. Alphs, Schizoph. Res, 2009, 113, 56–64 Search PubMed.
- L. Pani and G. Marchese, Expert Opin. Drug Delivery, 2009, 6, 319–331 Search PubMed.
- M. P. Jones, D. Nicholl and K. Trakas, Val. Health, 2008, 11, A580.
- H. S. Chow, Z. Chen and G. T. Matsuura, J. Pharm. Sci., 1999, 88, 754–758 CrossRef CAS.
- T. K. Vyas, A. K. Babber, R. K. Sharma, S. Singh and A. Misra, J. Pharm. Sci., 2006, 95, 570–580 CrossRef CAS.
- L. Li, I. Nandi and K. H. Kim, Int. J. Pharm., 2002, 237, 77–85 CrossRef CAS.
- Q. Zhang, X. Jiang, W. Jiang, W. Lu, L. Su and Z. Shi, Int. J. Pharm., 2004, 275, 85–96 CrossRef CAS.
- M. J. Lawrence and G. D. Rees, Adv. Drug Delivery Rev., 2000, 45, 89–121 CrossRef CAS.
- M. R. Patel, R. B. Patel, J. R. Parikh, K. K. Bhatt and A. J. Kundawala, Pharma. Rev., 2007, 5 Search PubMed . Available at: http://www.pharmainfo.net/reviews/microemulsions-novel-drug-delivery-vehicle. (Last accessed 08/08/2009).
- M. R. Patel, R. B. Patel, J. R. Parikh, A. B. Solanki and B. G. Patel, AAPS PharmSciTech, 2009, 10, 917–923 CrossRef.
- R. B. Patel, A. B. Patel, M. R. Patel, M. B. Shankar and K. K. Bhatt, Anal. Lett., 2009, 42, 1588–1602 CrossRef CAS.
- R. B. Patel, M. R. Patel, M. B. Shankar and K. K. Bhatt, J. AOAC Int., 2009, 92, 1082–1088 CAS.
- R. B. Patel, M. B. Shankar, M. R. Patel and K. K. Bhatt, J. AOAC Int., 2008, 91, 750–755 CAS.
-
Validation of Analytical Procedure: Methodology Q2 (R1) ( 1996) International Conference on Harmonization (ICH), International Federation of Pharmaceutical Manufacturers & Associations (IFPMA), Geneva, Switzerland Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.