Chromatographic analysis of major non-structural carbohydrates in several wood species – an analytical approach for higher accuracy of data
Michael
Raessler
*,
Bianka
Wissuwa
,
Alexander
Breul
,
Wolfgang
Unger
and
Torsten
Grimm
Max-Planck-Institut für Biogeochemie, Hans-Knoell-Str. 10, D-07745, Jena, Germany. E-mail: raessler@bgc-jena.mpg.de
First published on 23rd February 2010
Abstract
The exact and reliable determination of non-structural carbohydrates (NSC) in wood samples of different origin allows to gain further insight into the biogeochemical carbon cycle, i.e. processes that determine how carbon – and carbon dioxide – migrate between the land, the sea and the atmosphere. Plants are an essential part of this cycle using carbon dioxide in photosynthesis and locking up carbon within proteins and carbohydrates. Analytical determination of NSC is complicated by the complexity of the carbohydrate mixtures. However, most of them are present at the trace level only, and do not substantially contribute to the global NSC. Most important are glucose, sucrose, fructose and starch. Traditionally, these carbohydrates are analyzed spectrophotometrically. The spectrophotometric method does not provide any information on concentrations of sugar alcohols and raffinose. They can make up to a quarter of total NSC in deciduous tree samples. Consequently, ignoring them will severely impair the accuracy of NSC data in these samples. We will therefore present a method that allows the reliable determination of the following sugar alcohols and carbohydrates: inositol, sorbitol, mannitol, glucose, fructose, sucrose, raffinose and starch. These were the major contributors to the pool of non-structural carbohydrates in the samples we examined. It is important to note, that the portion of sugar alcohols and raffinose in the samples ranged from <2–25% of total NSC, irrespective of the plant species, with strongest impact (≥15% of NSC) for the conifer species “Pine” and the deciduous species “Ash Tree” and “Wild Cherry”. Application of our method substantially improved the accuracy of NSC data in these samples if compared to those of the spectrophotometrical analysis.
1. Introduction
The exact and reliable determination of non-structural carbohydrates (NSC) in plant samples of different origin allows to gain further insight into the biogeochemical carbon cycle,1,2i.e. processes that determine how carbon – and carbon dioxide – migrate between the land, the sea and the atmosphere. Plants are an essential part of this cycle using carbon dioxide in photosynthesis and locking up carbon within proteins and carbohydrates which represent the major form of photosynthetically assimilated carbon in the biosphere. Additionally, the exact determination of carbohydrate distribution may help better understand the seasonal behaviour of plant species, as carbohydrate composition varies with season, light availability and vegetative state.3,4NSC consists of various types of carbohydrates: sugar alcohols, such as inositol, sorbitol and mannitol, monosaccharides, such as glucose and fructose, disaccharides, such as sucrose, oligosaccharides, such as raffinose and the polysaccharide starch. With the exception of starch, these sugars are readily dissolved in polar solvents, and are therefore also referred to as “free carbohydrates”.
NSC have to be strictly distinguished from the structural carbohydrates which may comprise up to 75 wt% of vascular plant tissues,5 such as leaf, trunk, stem and root, but are also attached to the cell walls.6 In order not to over-estimate the NSC content of the sample, structural carbohydrates, i.e., cellulose, but also galacturonic acids and glycosylated arabinogalactan proteins, must be preserved. The absence of both arabinose and galactose in the examined extracts is therefore considered to indicate the conservation of structural carbohydrates.7 Consequently, both sugars were included in the analytical procedure and their chromatographic properties examined. Conservation of structural carbohydrates was performed by a prudent selection of the extraction conditions.3 In the present case, a hot water extraction helped release the carbohydrates and starch for NSC determination without affecting structural carbohydrates.
Despite the fact that NSC of wood samples consists of various types of carbohydrates, its determination is often carried out by UV spectrophotometry on microplates.4,8,9 This method, although easy to handle, is restricted to concentration data of glucose, fructose, sucrose and starch, only. No information on concentrations of sugar alcohols and raffinose will be provided. However, these compounds are often present in the samples at concentration levels that cannot be ignored if better accuracy of NSC data is desired. So far, the missing data have to be provided for by another analytical procedure; e.g., gas chromatography after derivatization.8 This is cumbersome and time-consuming. Additional sample preparation steps are necessary which may impair the integrity of the carbohydrate mixture in the sample. To overcome this insufficiency, a method is needed that readily provides the necessary data on all relevant NSC components, i.e. the sugar alcohols, glucose, fructose, sucrose, raffinose and starch without the need for additional and cumbersome sample manipulations. The results of our study will show that these compounds accounted for the overwhelming majority of NSC in the wood samples we examined. We will therefore consider them in the following, as “major non-structural carbohydrates”. Our method, based on isocratic high-performance anion-exchange chromatography with pulsed amperometric detection (HPAEC-PAD) using 18 mMol NaOH as eluent, allows the precise and sensitive determination of major NSC in less than 30 min.
PAD is well suited for sensitive carbohydrate analysis even in intricate matrices, such as plant extracts.10–12 Pulsed amperometric detection uses a triple-step potential waveform to combine amperometric detection with alternating anodic and cathodic polarizations to clean and reactivate the electrode surface. PAD has gained prominence as a selective and sensitive technique for the determination of carbohydrates due to the fact that oxide-free detection for aldehyde and alcohol-containing compounds at Au electrodes in alkaline media (pH 12) can be easily executed. The use of Au electrodes has the distinct advantage that detection can be achieved without simultaneous reduction of oxygen.
The method was carefully validated and successfully applied to different wood species: the conifers “Pine” and “Larch”, and the deciduous trees “Ash”, “Beech”, “Oak”, “Sessile Oak” and “Wild Cherry”. All the samples were also analyzed utilizing UV spectrophotometry on microplates to compare the results of the two methods. While there was a very good agreement of starch data, and of data representing the sum of glucose, fructose and sucrose, considerable deviations were observed for the “free carbohydrates”. Spectrophotometrical data did not include neither sugar alcohols nor raffinose, and were, therefore, distinctly lower than those obtained by HPLC-PAD. In fact, the chromatographical approach, providing the supplementary data on sugar alcohols and raffinose, yielded more accurate and reliable results of NSC determinations in these wood samples.
2. Materials and methods
2.1. Reagents and standards
NaOH 50%, HCl 30%, and HClO4 60%, suprapur, as well as fructose, galactose, glucose, sucrose (all p.A.) and soluble starch were purchased from Merck, Darmstadt, Germany. Arabinose and raffinose were purchased from Fluka, Buchs, Switzerland. Inositol, mannitol, and sorbitol were purchased from Sigma-Aldrich, Deisenhofen, Germany. Stock solutions of standards of each carbohydrate and sugar alcohol (1 g/L = 1000 ppm) as well as mixtures were prepared in ultra pure water, 18 Mohm (Millipore, Eschborn, Germany), and stored in 1 L plastic bottles at 4 °C in the refrigerator. Standard working solutions were prepared fresh daily prior to chromatographic analysis.2.2. Preparation of extracts
2.3. Chromatographic instrumentation
HPLC-PAD analyses were performed on a Dionex ICS 3000 ion chromatography system equipped with a gradient pump, a column oven and a pulsed amperometric detector. Electrochemical detection was achieved by use of the working pulse potentials and durations summarized in Table 1, all relative to a saturated Ag/AgCl reference electrode and a gold-working electrode.| Time (ms) | Potential (mV vs. Ag/AgCl) | Integration period |
|---|---|---|
| 0 | 100 | |
| 200 | 100 | Start |
| 400 | 100 | End |
| 410 | −2000 | |
| 420 | −2000 | |
| 430 | 600 | |
| 440 | −100 | |
| 500 | −100 |
Separation of carbohydrates and sugar alcohols was carried out on a CarboPac PA 10 (250 × 4.6 mm) analytical column which was preceded by a Carbo Pac PG 10 guard column. Both columns were operated at a constant temperature of T = 40 °C with 18 mMol NaOH as mobile phase. The analytical run lasted 26 min after which the column was flushed with 250 mMol NaOH for 10 min to prevent adsorption of carbonate and to remove strongly retained species. All eluents were constantly purged with helium to avoid formation of carbonate from aerial carbon dioxide. The sample injection volume was 25 μL.
2.4. Spectrophotometric analysis of glucose, fructose and sucrose
10 mg of fine ground wood material was boiled in 2 mL ultra pure water for 30 min. After centrifugation, an aliquot of 500 μL of the extract was treated with isomerase and invertase, to convert fructose and sucrose into glucose. The total amount of glucose was determined photometrically (λ = 340 nm) with a 96-well microplate photometer, HR 700, Hamilton, Reno, USA, after enzymatic conversion to gluconate-6-phosphate (hexokinase reaction). The remainder of the water extract was incubated with clarase for 15 h at 40 °C to break down starch to glucose. For details of the spectrophotometric method, see ref. 8,9.3. Results and discussion
3.1. Chromatographic separation of the selected carbohydrates and sugar alcohols
In the initial phase of our investigation, we were concerned with optimizing eluent composition, flow-rate and column flushing conditions in order to achieve maximum separation of the major non-structural carbohydrates in wood samples, while minimizing analysis time and variations in retention time. Baseline separation of inositol, sorbitol, mannitol, arabinose, galactose, glucose, sucrose, fructose and raffinose was obtained within 26 min, as indicated in Fig. 1. Stability of retention times after subsequent injections (N = 5) of standards was very good throughout (RSD <1% or better). In order to obtain reproducible retention times, it is essential that a post-run flush of the column with a strong NaOH solution be performed to wash strongly retained species.12 In the present case, the column was regenerated by elution with 250 mMol NaOH for 10 min before initial conditions were resumed and then equilibrated for 10 min with the initial mobile phase conditions prior to analysis. Good results for both standards and samples were obtained when maintaining the system in this manner.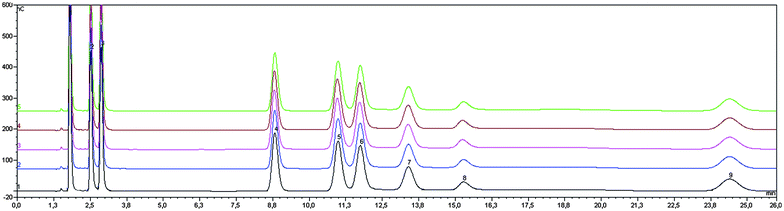 | ||
| Fig. 1 Overlay of chromatograms of standard mixture for determination of major NSC (N = 5) 1 inositol, 2 sorbitol., 3 mannitol, 4 arabinose, 5 galactose, 6 glucose, 7 sucrose, 8 fructose, 9 raffinose; concentration of each compound: 10 mg/L; flow 1 mL/min. | ||
3.2. Calibration ranges, detection limits and limits of quantification
For the carbohydrates the calibration range from 0.5–10 mg/L was checked for linearity. Calibration points were at 0.5, 1.0, 2.0, 4.0, 6.0, 8.0 and 10.0 mg/L. Linear regression analysis was used to calculate the slope, intercept and the correlation coefficient of each calibration curve which were constructed based on the peak area signals of each analyte. The detection limits, defined as signal to noise ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, were assessed as the minimum detectable quantity of each compound that could be detected without interference from the baseline noise. Calibration data were used for calculating the limits of detection (LOD) for each carbohydrate and sugar alcohol based on the absolute amount of standard injected onto the column using a 25 μL sample loop. LODs ranged from 9 pmol (mannitol) to 127 pmol (fructose, see Table 2). The limits of quantification (LOQ) accounted for the sample weight of 20 mg. LOQs ranged from 22 nmol (mannitol) to 317 nmol (fructose, see Table 2) and were, therefore, sufficiently sensitive for quantitative determination of carbohydrates and sugar alcohols in the wood extracts. In fact, LODs of this method were significantly lower than those recently reported for LC-MS studies on Lupinus albus extracts.13 Apart from LODs in [pMol] and LOQs both in [nMol] and [μg/L], Table 2 summarizes the regression equations and the regression coefficients R2 for the examined carbohydrates and sugar alcohols.
:1, were assessed as the minimum detectable quantity of each compound that could be detected without interference from the baseline noise. Calibration data were used for calculating the limits of detection (LOD) for each carbohydrate and sugar alcohol based on the absolute amount of standard injected onto the column using a 25 μL sample loop. LODs ranged from 9 pmol (mannitol) to 127 pmol (fructose, see Table 2). The limits of quantification (LOQ) accounted for the sample weight of 20 mg. LOQs ranged from 22 nmol (mannitol) to 317 nmol (fructose, see Table 2) and were, therefore, sufficiently sensitive for quantitative determination of carbohydrates and sugar alcohols in the wood extracts. In fact, LODs of this method were significantly lower than those recently reported for LC-MS studies on Lupinus albus extracts.13 Apart from LODs in [pMol] and LOQs both in [nMol] and [μg/L], Table 2 summarizes the regression equations and the regression coefficients R2 for the examined carbohydrates and sugar alcohols.
| Carbohydrate | Regression equation | R 2 | LOD [pMol] | LOQ [nMol] | LOQ [μg/L] |
|---|---|---|---|---|---|
| Inositol | y = 0.714x + 0.137 | 0.9988 | 22 | 55 | 10 |
| Sorbitol | y = 0.518x + 0.159 | 0.9993 | 17 | 42 | 8 |
| Mannitol | y = 0.614x + 0.073 | 0.9998 | 9 | 22 | 4 |
| Arabinose | y = 0.402x + 0.302 | 0.9995 | 59 | 147 | 22 |
| Galactose | y = 0.677x + 0.280 | 0.9998 | 33 | 82 | 15 |
| Glucose | y = 0.727x + 0.455 | 0.9994 | 55 | 137 | 25 |
| Sucrose | y = 0.469x − 0.081 | 0.9993 | 28 | 70 | 24 |
| Fructose | y = 0.235x + 0.182 | 0.9966 | 127 | 317 | 57 |
| Raffinose | y = 1.020x + 0.035 | 0.9991 | 22 | 55 | 28 |
3.3. Extraction of carbohydrates and sugar alcohols
The extraction procedure is vital for the accurate determination of non-structural carbohydrate composition. Total sugar extracted will considerably depend on the method chosen.14 The higher the polarity of the solvent, the more readily the carbohydrates will be dissolved. However, acids, even when diluted, cannot be used. They cause both the dissolution of structural carbohydrates and the hydrolysis of di- and oligosaccharides. On the other hand, Raessler et al.,15 reported good efficiency of water for the extraction of fructans and inulin in rye grass samples. Additionally, water is highly compatible with the chromatographic procedure raising no concern of solvent interferences. Both the efficiency (i.e. completeness) and the reliability (i.e. reproducibility, no dissolution of structural carbohydrates) of the extraction procedure were checked by the use of a real sample: “Pine”. The same sample was also used for checking starch extraction and hydrolysis (cf. section 3.4.). The efficiency was checked by a careful repetition of the whole extraction procedure: after centrifugation, the water phase containing the “free carbohydrate” fraction was removed, and 5 mL of ultra pure water were added to the residual pellet which was placed to the shaking water bath at T = 80 °C for another 30 min.This second water phase was separately analyzed by HPLC-PAD as described under 3.1. The chromatogram revealed no carbohydrates which confirmed the efficiency of the extraction procedure. To check the reliability, six subsequent extractions of the “Pine” sample were carried out and analyzed. The overlay of the six chromatograms of each extract of the “Pine” sample is shown in Fig. 2a. As can be seen, highly consistent results for the identified sugar alcohol (inositol) and carbohydrates (glucose, fructose, sucrose) were obtained (RSD values < 5%). Identification of compounds was carried out by comparison with the standard mixture. Additionally, there was almost no arabinose and galactose present in the extracts which indicated the preservation of structural carbohydrates, thus confirming the suitability of the extraction procedure for reliable NSC determinations.7
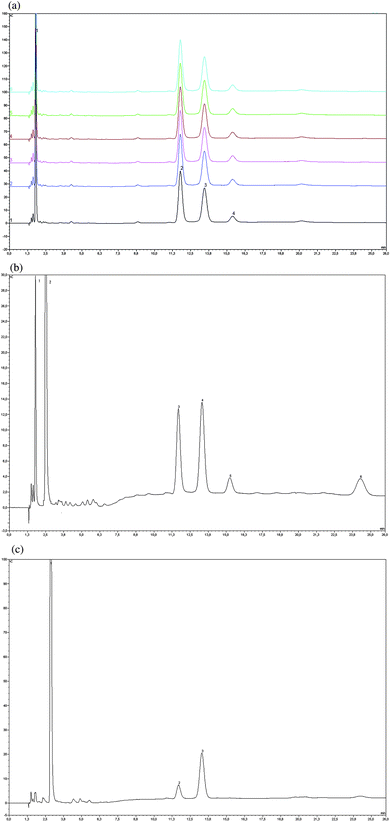 | ||
| Fig. 2 a Overlay of chromatograms of extract of “Pine” sample (N = 6) 1 inositol, 2 glucose, 3 sucrose, 4 fructoseb Chromatogram of extract of “Wild Cherry” sample 1 inositol, 2 sorbitol, 3 glucose, 4 sucrose, 5 fructose, 6 raffinosec Chromatogram of extract of “Ash Tree” sample 1 mannitol, 2 glucose, 3 sucrose. | ||
3.4. Extraction and hydrolysis of starch
The accurate determination of the high molecular weight polysaccharide starch relies on both its complete extraction from the sample, and its complete hydrolysis into the monomer glucose; the latter being used for analytical quantification of starch. In plant samples, starch granules are often included in a protein layer within the cell walls, which considerably limits the accessibilty of the solvent during extraction. Consequently, extraction of starch generally needs rather harsh conditions to allow thorough access of the solvent. The process fostering the dissolution of starch, is called dispersion. It is favored by disruption of the cell walls. This is efficiently achieved by sonomechanical energy, i.e. ultrasonic radiation.16–18 The ultrasonic effects on cells are based on the collapse of cavitation bubbles microstreaming near the boundary layer, by which cell membranes are fragmented. Due to the more efficient dispersion, sonification-assisted extraction should result in higher yields of starch than obtained by extracting the sample on a shaking water bath as utilized for sugar alcohols and soluble carbohydrates (cf. section 3.3.). We therefore examined the starch extraction efficiency of both methods: the “Pine” sample was submitted to the following extraction procedures: ultrasonic water bath at T = 80 °C (power 720 W) for 30 (A) and 60 (B) min; shaking water bath at T = 80 °C for 30 (C) and 60 (D) min, respectively. Each procedure was carried out in duplicate. As expected, ultrasonic-assisted extraction resulted in considerably higher starch yields compared to shaking water bath extractions: A: 6.5 ± 0.8 [g/kg], B: 7.0 ± 0.5 [g/kg]; C: 4.5 ± 0.8 [g/kg], D: 5.1 ± 0.6 [g/kg]. Although the influence of duration of exposure to ultrasonic energy on starch yield was rather small, we referred to the 60 min term for starch extraction of our samples. Starch yield was determined after acidic hydrolysis. Hydrolysis is a necessary step prior to analytical determination of high molecular weight polysaccharides. It can be performed either enzymatically or by the use of acids. Acidic hydrolysis is much faster compared to the more time-consuming incubation of enzymes, which needs up to 15 h.4,8 Moreover, highest efficiency of enzymes is restricted to a narrow pH range only, which often demands buffering of the solution. Addition of buffer salts, however, can impair the chromatographic separation, giving raise to shifts of retention times. While insufficient recovery rates were reported in case diluted acids were used for starch hydrolysis,4 much better results were obtained utilizing semi-concentrated perchloric acid.19 We therefore examined the hydrolytic power of HClO4 30%. 5 mL of HClO4 60% were added to the same volume of a standard solution containing 10 mg/L of starch in water. This solution was exposed to a water bath at T = 80 °C for 60 min. The hydrolysate was afterwards treated as described in section 2.2. and glucose determined by HPLC-PAD. Mass balances established after 10 separate starch hydrolyses revealed a glucose recovery of 97 ± 5%, taking account of the hydrolytic equilibrium of the species in solution. Starch concentration in the conifer samples “Pine” and “Larch” were rather low (7 g/kg), but considerably higher in the deciduous tree samples (up to 168 g/kg, cf.Table 3). Appropriate dilution of the hydrolysates made sure that the resulting glucose concentrations remained within the calibration range. Additionally, dilution helped stabilize the chromatographic determination of glucose. The perchlorate concentration was sufficiently reduced not to affect the signal characteristics of the glucose peak (retention time, signal intensity, peak shape). Ions with affinity to the anion-exchange column, can impair the analytical procedure.20 The accuracy of the starch results obtained in this way was checked by comparing them with the results of the spectrophotometrical analysis on microplates after enzymatic hydrolysis (“hexokinase-method”, cf. section 3.6. for details).| Sample | Inositol | Sorbitol | Mannitol | Glucose | Sucrose | Fructose | Raffinose | Starch | NSC |
|---|---|---|---|---|---|---|---|---|---|
| Ash Tree | 21.3 | 1.3 | 6.8 | 57 | 86.4 | ||||
| Beech Tree | 2.3 | 2.3 | 13.3 | 3.8 | 0.7 | 168 | 190.4 | ||
| W. Cherry | 2.5 | 5.6 | 0.6 | 5.3 | 0.9 | 1.6 | 45 | 61.5 | |
| Larch | 1.3 | 4.3 | 6 | 5.3 | 0.4 | 7 | 24.3 | ||
| Oak Tree | 9.8 | 7.2 | 9.2 | 11.4 | 8.1 | 73 | 118.7 | ||
| S. Oak | 9.8 | 14.3 | 5.7 | 18.9 | 115 | 163.7 | |||
| Pine | 4.8 | 7 | 8.3 | 4 | 7 | 31.1 |
3.5. Analyses of wood extracts
After the suitability and the reliability of the method had been demonstrated by utilizing the “Pine” sample, other wood species were submitted to the analytical procedure and their composition of major non-structural carbohydrates examined: the conifer “Larch”, and the deciduous trees “Ash”, “Beech”, “Oak”, “Sessile Oak”, “Wild Cherry”. As with the “Pine” sample, identification of carbohydrates in the wood extracts was achieved by comparison of peak retention times with those of the standards. Due to careful rinsing and re-equilibration of the column between each analytical run, retention times of the carbohydrates and sugar alcohols in the extracts remained highly stable. No addition of an internal standard was necessary which considerably simplified sample preparation. Carbohydrate composition of the “Pine” sample has already been discussed in section 3.3 together with the studies on the reliability of the extraction procedure. The overlay of the chromatograms of six separately analyzed extracts, shown in Fig. 2a, revealed presence of the sugar alcohol inositol and the carbohydrates glucose, sucrose and fructose. A similar pattern revealed the chromatogram of the “Larch” sample (not shown), including traces of raffinose (0.4 g/kg). The two conifer samples were characterized by a homogenous distribution of carbohydrates. Contrary to the deciduous tree species, where starch was by far the dominant contributor to NSC (≥ 65% of NSC in these samples were made of starch), portion of starch in the conifer species was comparable to that of glucose, sucrose and fructose. Portion of sugar alcohols and raffinose, however, was smaller ranging from 7% (“Larch”) to 15% (“Pine”). With the exception of the “Ash Tree” and “Sessile Oak”, presence of the oligosaccharide raffinose was more pronounced in deciduous tree samples: while significantly lower than either glucose or fructose in the “Beech Tree” (0.7 g/kg), it equalized the amount of glucose and almost of sucrose in the “Oak Tree” (8.1 g/kg, which was by far the highest absolute raffinose concentration found). Although the raffinose amount was much lower in the “Wild Cherry” (1.6 g/kg), it surpassed that of both glucose and fructose. These results emphasized the presence of raffinose as a physiologically important carbohydrate whose maximum concentrations are transported in the phloem sap of several plant species in the cold seasons to improve their winter hardiness.21The “Wild Cherry” revealed another interesting feature: it showed the highest diversity of sugar alcohols (inositol, sorbitol) and carbohydrates (glucose, sucrose, fructose and raffinose) of all examined samples (cf.Fig. 2b). Despite the low concentrations of glucose and fructose (<1 g/kg, see Table 3), their peaks were distinctly separated from the baseline. Due to the low limits of quantification of the method, both compounds were unambiguously identified and quantified. Considerably higher concentrations were observed for the sugar alcohols inositol and sorbitol (in total 8.1 g/kg). Portion of sugar alcohols and raffinose in the “Wild Cherry” was 16% of NSC and therefore higher than the sum of glucose, sucrose and fructose in this sample (11%). The impact of sugar alcohols on the exact determination of major NSC was most pronounced in the “Ash Tree”. A huge peak at 2.9 min referred to the sugar alcohol mannitol; two smaller ones to glucose and sucrose, respectively (cf.Fig. 2c). Quantitative evaluation of the mannitol peak resulted in a concentration of 21.3 g/kg; almost three times the sum of glucose and sucrose; representing a quarter of non-structural carbohydrates in this sample.
These two examples clearly demonstrated that sugar alcohols and raffinose were important contributors to NSC in wood extracts. With the exception of the “Beech Tree” and “Sessile Oak”, portion of sugar alcohols and raffinose in the deciduous tree species was ≥10%, an impact too high to be ignored. In case, only “free carbohydrates”, i.e. all relevant carbohydrates, except starch, were considered, their portion raised to a maximum of 70%. A reliable and accurate analysis of NSC cannot renounce at their identification and quantification. This happened, however, when samples were analyzed spectrophotometrically. Consequently, substantial differences were observed when results of the two methods were compared (for details, see section 3.6).
Apart from the identified peaks of inositol, sorbitol, glucose, sucrose, fructose and raffinose (“Wild Cherry”, Fig. 2b) and mannitol, glucose, sucrose (“Ash Tree”, Fig. 2c), both chromatograms revealed a number of tiny peaks close to the elution of sugar alcohols and in the retention time region between 3.8 and 6 min. If at all, these peaks reflected compounds at extremely low concentration levels, which we considered not to affect accuracy of NSC results significantly. This was also true for the other wood samples (“Larch”, “Beech”, “Oak Tree” and “Sessile Oak”) whose chromatograms are not shown. Significant contributions to non-structural carbohydrates in these samples were solely made by the sugar alcohols and carbohydrates listed in Table 3. This table summarizes their presence and concentrations (in g/kg) for each sample. As mentioned above, starch concentrations in the conifer species (7 g/kg) differed markedly from those of the deciduous species (maximum at 168 g/kg; “Beech Tree”). If appropriately diluted, analyses of samples with high amounts of starch caused no problems. Starch results obtained by HPLC-PAD after acidic hydrolysis were in good agreement with those of spectrophotometrical analyses (cf. next section).
The individual composition of NSC (in percent sugar alcohols & raffinose, glucose, sucrose, fructose and starch) for each sample is shown graphically in Fig. 3a–g.
![a “Ash Tree”; composition of NSC in [%] b “Beach Tree”; composition of NSC in [%] c “Wild Cherry”; composition of NSC in [%] d “Larch”; composition of NSC in [%] e “Oak Tree”; composition of NSC in [%] f “Sessile Oak”; composition of NSC in [%] g “Pine”; composition of NSC in [%].](/image/article/2010/AY/b9ay00193j/b9ay00193j-f3.gif) | ||
| Fig. 3 a “Ash Tree”; composition of NSC in [%] b “Beach Tree”; composition of NSC in [%] c “Wild Cherry”; composition of NSC in [%] d “Larch”; composition of NSC in [%] e “Oak Tree”; composition of NSC in [%] f “Sessile Oak”; composition of NSC in [%] g “Pine”; composition of NSC in [%]. | ||
Finally, absence of both arabinose and galactose in the chromatograms confirmed that no structural carbohydrates were affected and strengthened the reliability of our NSC data.
3.6. Comparison with the spectrophotometric method after enzymatic hydrolysis
All samples were also analyzed utilizing spectrophotometry after enzymatic hydrolysis, as described in section 2.4. Starch was broken down to glucose after incubation of invertase. The amount of glucose was determined at λ = 340 nm with a 96-well photometer. The results obtained in this way were in good agreement with those of HPLC-PAD. Statistical correlation of starch data yielded a regression equation y = 1.16x + 0.37 with a correlation factor R2 = 0.993. This emphasized that starch analysis by HPLC-PAD after hydrolysis with HClO4 30% could be considered a valuable alternative to the traditional method, in case the time-consuming incubation of enzymes is not desired. Additionally, due to its higher sensitivity, HPLC-PAD will also allow the reliable determination of trace amounts of starch that otherwise could not be detected. In another step, the sum of glucose, sucrose and fructose was determined spectrophotometrically, based on the hexokinase reaction (sugar alcohols and raffinose were not amenable to the spectrophotometrical analysis). For this purpose, sucrose was enzymatically split into glucose and fructose by the use of invertase; fructose being then converted into glucose using the enzyme isomerase. The glucose was finally analyzed as gluconate-6-phosphate. This glucose amount was compared to the corresponding concentrations measured by HPLC-PAD. These data correlated well (y = 0.96x + 0.14; R2 = 0.992) If, on the other hand, these spectrophotometrical data were compared to the concentrations of “free carbohydrates” (i.e. glucose, sucrose, fructose, as well as sugar alcohols and raffinose), determined by HPLC-PAD, substantial differences of results were observed: “Ash Tree” (HPLC 2.7; UV 1.0% DM); “Wild Cherry” (HPLC 1.6; UV 0.8% DM); “Oak Tree” (HPLC 4.5; UV 2.5% DM), which led to a worse correlation of data (y = 0.65x + 0.24; R2 = 0.82). As expected, spectrophotometrical analysis of samples with substantial concentrations of sugar alcohols and/or raffinose inevitably resulted in an under-estimation of the “free carbohydrate” portion. Although spectrophotometrical analyses provided good quality results for starch and the sum of glucose, sucrose and fructose, the outcome of these studies clearly demonstrated the superiority of our method for a more accurate determination of major non-structural carbohydrates, taking account of the importance of sugar alcohols and raffinose.4. Conclusion
It could be shown, that the contribution of sugar alcohols and raffinose, which can make up a quarter of the pool of non-structrural carbohydrates of several tree species, such as “Ash Tree”, “Wild Cherry”, “Oak Tree” or “Pine”, is important and cannot be ignored if a high degree of accuracy of analytical data is desired. These compounds are not recorded by the traditional spectrophotometrical determination. To fill the gap, the missing data have to be provided for by having recourse to additional analytical devices. Our method, based on rugged and easy-to-handle chromatography, allows the individual determination of the sugar alcohols inositol, mannitol, sorbitol, the monosaccharides glucose, fructose, and the oligosaccharides sucrose and raffinose in a single run. Together with the polysaccharide starch, which is determined as glucose after acidic hydrolysis, all major components of non-structural carbohydrates are determined with the same instrumentation using the same chromatographic conditions. There is base-line separation of all relevant compounds within less than 30 min. The separations being characterized by a high degree of stability and reproducibility. There is no need for elaborate sample preparation (e.g. addition of an internal standard). Due to its versatility and sensitivity, it can easily be extended to the analysis of other carbohydrates – at varying concentration levels – according to the analytical problem. The presented method is a substantial contribution for a more accurate determination of non-structural carbohydrates in wood species.Acknowledgements
The authors are very grateful to Dr Günter Hoch, Institute of Botany, University of Basel, Switzerland, for carrying out the spectrophotometric analyses of the wood samples in his laboratory and for fruitful comments and discussions.5. References
- M. A. Teace and M. L. Fogel, Org. Geochem., 2007, 38, 458–473 CrossRef.
- M. Raessler, Proceedings of the 33rd International Symposium on High Performance Liquid Phase Separations and Related Techniques (HPLC 2008), Kyoto, Japan, Dec 2–5, 2008, p. 101 Search PubMed.
- L. Gomez, M. O. Jordan, S. Adamowicz, H. Leiser and L. Pagès, Cah. Agric., 2003, 12, 369–386 Search PubMed.
- P. S. Chow and M. S. Landhäusser, Tree Physiol., 2004, 24, 1129–1136 Search PubMed.
- P. M. Medeiros, M. H. Conte, J. C. Weber and B. R. T. Simoneit, Atmos. Environ., 2006, 40, 1694–1705 CrossRef CAS.
- S. Willför, A. Pranovicha, T. Tamminen, J. Puls, C. Laine, A. Suurnäkki, B. Saake, K. Uotila, H. Simolin, J. Hemming and B. Holmbom, Ind. Crops Prod., 2009, 29, 571–580 CrossRef.
- M. Raessler, Proceedings of Euroanalysis 2009, Innsbruck, Austria, Sep 6–10, 2009, p. 94 Search PubMed.
- G. Hoch, A. Richter and C. Körner, Plant, Cell Environ., 2003, 26, 1067–1081 CrossRef CAS.
- S. C. Wong, Photosyn. Res., 1990, 23, 171–180 CrossRef CAS.
- W. R. LaCourse, Pulsed electrochemical detection of carbohydrates at noble metal electrodes following liquid chromatographic and electrophoretic separation, in Carbohydrate Analysis by Modern Chromatography and Electrophoresis, Ed. Z. W. Rassi, Journal of Chromatography Library, Elsevier Science B.V., 2002, Vol. 66, 905–946 Search PubMed.
- T. R. I. Cataldi, C. Campa and G. E. DeBenedetto, Fresenius J. Anal. Chem., 2000, 368, 739–758 CrossRef CAS.
- D. Blanco, D. Muro and M. D. Gutiérrez, Anal. Chim. Acta, 2004, 517, 65–70 CrossRef CAS.
- C. Antonio, P. Pinheiro, M. M. Chaves, C. P. Ricardo, M. F. Ortuno and J. T. Oates, J. Chromatogr., A, 2008, 1187, 111–118 CrossRef CAS.
- F. Davis, L. A. Terry, G. A. Chope and C. F. Faul, J. Agric. Food Chem., 2007, 55, 4299–4306 CrossRef CAS.
- M. Raessler, B. Wissuwa, A. Breul, W. Unger and T. Grimm, J. Agric. Food Chem., 2008, 56, 7649–7654 CrossRef CAS.
- Z. Hromadkova and A. Ebingerova, Ultrason. Sonochem., 2003, 10, 127–133 CrossRef CAS.
- L. Wang and Y. J. Wang, J. Cereal Sci., 2004, 39, 291–296 CrossRef CAS.
- Q. Z. Zhang, A. Kullmann and J. M. Greef, J. Agron. Crop Sci., 1992, 169, 217–222 CrossRef CAS.
- F. R. Humphreys and J. Kelly, Anal. Chim. Acta, 1961, 24, 66–70 CrossRef CAS.
- C. Panagiotopoulos and R. Sempéré, Limnol. Oceanogr. Methods, 2005, 3, 419–454 Search PubMed.
- N. Pavis, N. J. Chatterton, P. A. Harrison, S. Baumgartner, W. Praznik, J. Boucaud and M. P. Prud'-homme, New Phytol., 2001, 150, 83–95 Search PubMed.
| This journal is © The Royal Society of Chemistry 2010 |