Rapid and automated sequential determination of ultra-trace long-lived actinides in air filters by inductively coupled plasma mass spectrometry
Dominic
Larivière
a,
Karima
Benkhedda
*b,
Stephen
Kiser
b,
Sonia
Johnson
b and
R. Jack
Cornett
b
aLaboratoire de Radioécologie, Département de chimie, Université Laval, 1045 Avenue de la Médécine, Québec, QC, Canada G1V 0A6
bKarima Benkhedda, Radiation Protection Bureau, 775 Brookfield Road, Address Locator 6302D1, Ottawa, ON, Canada K1A 1C1. E-mail: karima_benkhedda@hc-sc.gc.ca; Fax: (613) 957-1089; Tel: (613) 957-9039
First published on 15th January 2010
Abstract
A novel, rapid and completely automated method for the determination of long-lived actinides in air particulate samples was developed using ion chromatography separation prior to ICP-MS analyses. U, Pu and Am were pre-concentrated and separated from radioactive and stable interferences through a sequential arrangement of three columns (TEVA, U/TEVA, DGA) arranged in order to allow selective retention of the radioisotope on the designated resin. Detection and quantification of the various radioisotopes were performed on-line using inductively coupled plasma mass spectrometry (ICP-MS). Detection limits of 0.0006 (238U), 0.0063 (239Pu), 0.0041 (240Pu), 0.062 (241Am) μBq/m3 were obtained for air filters (based on 3000 m3 of air filtered). The automated procedure has been successfully tested on SRMs and spiked samples and showed high recovery yields (>90%). A total analysis time of 18 min is required for the separation, detection, and column rinsing/regeneration for subsequent analysis.
Introduction
The determination of long-lived isotopes of actinides such as uranium, plutonium, and americium in the biosphere is important as it is linked to the use of nuclear technologies. This connection makes them valuable indicators of nuclear activities worldwide.1–3 In addition, in the aftermath of a radiological/nuclear (R/N) event they are among the critical radioisotopes used to articulate an effective consequence management response.4–7 Measurement of the level of environmental contamination should be performed for every environmental compartment (e.g. water, soil, vegetation) to completely assess the extent of the contamination. While comprehensive global monitoring should be performed to detect releases, airborne contamination should be considered as the most critical compartment as it leads to significant radiation dose, following environmental dispersion, to the lungs if the airborne radionuclides are inhaled.8–10 Air is also a critical medium as dispersion of materials is largely influenced by factors such as wind and precipitation. Other possible sources of internal contamination, namely food and water, can be rapidly replaced with non-contaminated supplies, unlike the situation of airborne contamination.8,9 Therefore, analytical methods used to detect the presence of radiological airborne contamination, especially those applied to evaluate the impact of a radiological release, are expected to have both radioelemental selectivity and multi-isotopic capabilities while providing the analytical results in a timely manner.4–7Alpha-spectrometry has been frequently applied to the determination of long-lived α-emitters. However, the fact that many of the isotopes of Pu, Am, and U have low-specific activity indicate that either large amount of samples will be required or that longer counting time will be needed for adequate detection.11–13 In addition, separation protocols used for α-spectrometry samples are generally labor intensive, time-consuming, and focused on the separation of only one radioelement to produce thin chemically pure sources free of energy interferences.1,4,5,7,14 For all these reasons, α-spectrometry is not perfectly suited for emergency response. In comparison, accelerator mass spectrometry (AMS)1,15–17 and thermal ionization mass spectrometry (TIMS),17–19 are exceptionally sensitive ion-counting methods that can detect a variety of long-lived actinides down to the sub-fg range. However, the scarcity of those instruments, especially AMS, and their low sample throughput make them less attractive instrumentation for emergency response scenarios.20,17 Inductively coupled plasma mass spectrometry (ICP-MS), while less sensitive than the above-mentioned techniques17 but far more common in analytical laboratories, has demonstrated its effectiveness for the measurement of many long-lived radionuclides at higher environmental concentrations.20–23 The simplicity of the interface of the ICP, which allows coupling with a variety of analytical techniques, including a multi-solvent delivery system (MDS) such as high-performance liquid chromatography on ion-exchange pumps, and its multi-elemental ability are attractive features to develop methods that provide significant sample throughput without sacrificing instrumental sensitivity.20
Many protocols have demonstrated the analytical capability of ICP-MS for the on-line determination of specific long-lived radioelements in a variety of matrices such urine24–27 water28–30 soil, sediment,3,31–34 biological3,33 and vegetation35 samples and food.36,33 However, most of these protocols lack the multi-elemental capacity and rapidity required for post-event assessment. The ones that have embraced the multi-elemental capacity of the ICP-MS have focused primarily on nuclear fuels and wastes quantification2,37–40 While these methods provide selectivity, rapidity, and a certain degree of automation, they lack the sensitivity required for environmental radioactivity monitoring, which can be achieved only through sample pre-concentration5,7
In this work and based on the knowledge available for the automation of nuclear waste analysis, we have investigated and demonstrated the automation of a sequential injection system for the identification and quantification of airborne actinides. The automated system is composed of a multi-solvent delivery system for pre-concentration/separation coupled to a sector-field ICP-MS (ICP-SFMS) for on-line detection. The determination of 238U, 239,240,242Pu, and 241,243Am in certified materials (airborne particulate matter and loaded air filters) and spiked filters was performed using the method developed. Parameters such as interferences from uranium hydride, abundance sensitivity from 238U peaks, matrix effect, and instrumental sensitivity have been investigated and method performance criteria such as detection limits, sample throughput, accuracy and precision are also reported.
Experimental
Instrumentation
All measurements were performed on an Element2 ICP-SFMS (Thermo Fisher Scientific, Bremen, Germany). A high sensitivity inlet system (Apex-HF, Elemental Scientific Inc., Omaha, NE) coupled to a microflow perfluoroalkoxy (PFA) nebulizer was used as sample introduction system. Instrumental conditions for the Element2 ICP-SFMS and the Apex-HF were set to ensure high instrumental sensitivity and good precision during the measurement (Table 1).| Instrumental parameters | Element2 ICP-SFMS |
| Torch position | Optimized daily |
| Gas flow (L/min) | |
| Cooling | 16.08 |
| Auxiliary | 0.81 |
| Sample | 0.99 |
| RF power (W) | 1200 |
| Lenses (V) | |
| Extraction | −2000 |
| Focus | −902 |
| X-deflection | 0.00 |
| Shape | 120.00 |
| Y-deflection | −3.8 |
| Detector voltage (V) | 1600 |
| Sampling cone | 1.1 mm Nickel |
| Skimmer cone | 0.8 mm Nickel |
| m/z monitored | 235, 238, 239, 240, 241, 242, 243 |
| Number of passes | 1 |
| Number of replicates | 350 |
| Acquisition time (min) | 7 |
| Apex-Q system parameters | |
|---|---|
| Nebulizer | 1 mL/min PFA microflow |
| Spray chamber temperature (°C) | 140 |
| Peltier-cooled multipass condenser temperature (°C) | −5 |
A multi-solvent delivery system (ICS-3000, Dionex, Sunnyvale, CA) was used to deliver the various solvents in a gradient mode. An autosampler, (AS-HV, Dionex, Sunnyvale, CA) was used for the automated sample loading through the columns. The direction of the solvent flow into the column was controlled using four biocompatible analytical-scale two-positions, six-port switching modules (MX9900-000, Upchurch Scientific, Oak Habor, WA). Biocompatible modules were used instead of stainless steel ones to reduce the risk of contamination as well as deterioration due to corrosion when using acid solutions. The ICS-3000 unit, the switching modules and the autosampler were all controlled by the CHROMELEON® chromatography data system (Dionex, Sunnyvale, CA). Fig. 1 is a schematic diagram of the setup of the system coupled to the ICP-SFMS. Isotemp® programmable muffle furnace (Fisher Scientific, Ottawa, ON) and a MARS 5 microwave system (CEM Corporation, Matthews, NC, USA) were used for the thermal destruction and the acid digestion of the samples, respectively.
 | ||
| Fig. 1 Schematic of the HPLC-ICP-MS system for the sequential separation and detection of actinides. | ||
Reagents and materials
High-purity water (18 MΩ/cm) prepared by a Milli-Q® ultrapure water purification system (Millipore, Bedford, MA) was used throughout this work. Optima grade concentrated HNO3 (Fisher Scientific, Ottawa, ON) was used for sample digestion and to prepare working solutions and standards. A 0.1 mol L−1 oxalate solution was prepared by dissolution of the appropriate amount of ACS-certified ammonium oxalate monohydrate salt (Fisher Scientific, Ottawa, ON) in Milli-Q water. 239,240,242Pu (SRM-4330B, -4338A, and -4334G, respectively) and 241,243Am (SRM-4322B and -4332D, respectively) used as spikes were purchased from NIST (Gaithersburg, MD). Uranium standard solutions were prepared from 1000 μg mL−1 Claritas PPT®-grade standard (Spex CertiPrep, Metuchen, NJ). Working solutions were prepared by serial dilution of U, Pu, and Am standards in 3 mol L−1 HNO3.Three stainless steel analytical grade columns (Alltech, Columbia, MD), two 4.6 mm i.d. × 50 mm long and one 2.1 mm i.d. × 50 mm long, coated inside with polyether ether ketone (PEEK) were filled, respectively, with TEVA,41 U/TEVA,42 and n-DGA43 resins (50–100 μm particle size, Eichrom Technologies Inc., Darien, IL). All transport and reagent lines used to design the flow injection (FI) unit, were made of 0.762 mm i.d. PEEK tubing (Alltech, Columbia, MD) with the exception of the transfer line between the switching module four and the PFA nebulizer which was assembled using 1.1 mm i.d. polytetrafluoroethylene (PTFE) tubing (Alltech, Columbia, IL). 10–32 PEEK high pressure fittings with PEEK ferrules (Upchurch Scientific, Oakhabor, WA) for coned ports were used to connect the switching modules, multisolvent delivery system, and the analytical-grade columns.
Reference materials
Two reference materials, SRM-1648 (atmospheric urban particulate matter) and SRM-2783 (air particulate matter on polycarbonate filter media) from the National Institute of Standards and Technology (NIST, Gaithersburg, MD) were used to validate the analytical protocol developed for the determination of U, Pu, and Am after sample preparation and preconcentration. These materials were chosen to mimic composition of dust collected in air filters. Since only uranium content is certified in these materials, known amounts of 239,240,242Pu and 241,243Am were spiked into the samples to assess analyte recovery during sample preparation and analysis.In addition, in order to assess the efficiency of the sample preparation and analyte detection developed in this work on actual air filters currently used by the Canadian Radiological Monitoring Network, polypropylene (PP) 8′′x 10′′ (20.5 × 25.4 cm) filters, (3M, London, ON) were spiked with known amounts of the radioisotopes (U, 239,240,242Pu and 241,243Am) and their recovery, following sample preparation and analysis, was evaluated.
Flow injection (FI) system
Detailed operation of the automated system (with the exception of loading/reloading step) is described in Table 2. Although the proposed protocol lasts only 16 min, the next sample can only be injected 18 min after the first injection, since one minute is required for the CHROMELEON® software to process the data, and another one to stabilize the flow rate. Three of the switching valves in the system are used for the sequential extraction of Pu, U, and Am, while the last module is designed to either direct the sample or a rinsing solution (2% HNO3) to the nebulizer/ICP-MS.| Step | Time (s) | Medium | Flow rate (mL/min) | Switching modules (SM) positiona | ||||
|---|---|---|---|---|---|---|---|---|
| SV-1 | SV-2 | SV-3 | SV-4 | SLPb | ||||
| a SV-1 to SV-4: switching valves. b Sample loop position, elute signifies that the medium is passing through the sample loop while load is the opposite. | ||||||||
| 1 | 240 | 3M HNO3 | 2.5 | On | On | On | Off | elute |
| 2 | 270 | 0.1M (NH4)2C2O4 | 1 | Off | On | Off | On | load |
| 3 | 120 | 0.1M (NH4)2C2O4 | 1 | Off | Off | On | On | load |
| 4 | 180 | 0.01M (NH4)2C2O4 | 1 | On | Off | Off | On | load |
| 5 | 90 | Milli-Q water | 2.5 | On | On | On | Off | load |
| 6 | 60 | 3M HNO3 | 3 | On | On | On | Off | load |
| Step | Step description |
|---|---|
| 1 | 3 M HNO3 is pumped through the sample loop to load the sample and rinse the residual elements from the three resins |
| 2 | 0.1 M (NH4)2C2O4 is pumped through U/TEVA to elute U |
| 3 | 0.1 M (NH4)2C2O4 is pumped through n-DGA to elute Am |
| 4 | 0.01 M (NH4)2C2O4 is pumped through TEVA to elute Pu |
| 5 | Milli-Q water pumped to rinse all three resins from any residual elements |
| 6 | 3 M HNO3 is pumped through all three resins to pre-condition the resins for the next analysis |
Sample preparation
Polypropylene filters (in-house filters) and 0.2 g (in triplicates) of urban particulate matter samples (SRM-1648) were ashed at 550 °C for 4 h in porcelain crucibles. The SRM-2783 reference material substrate was made of polycarbonate and therefore did not require an ash step. After cooling, some of the PP filters were spiked with 5 μg L−1 of U, 2.52 Bq L−1 of 239,240Pu and 241Am, 0.14 Bq L−1 of 242Pu and 1 Bq L−1 of 243Am and the urban particulate matter samples were spiked with the same amount of Pu and Am but no U was added. The residual ash was dissolved with 6 mL of concentrated HNO3 and transferred into a microwave vessel and was digested with 2 heating steps at 1600 W: ramping to 200 °C in 10 min and staying at 200 °C for 30 min. Non-spiked filters were used to monitor the levels of these actinides in the filters and to evaluate method detection limit for each radionuclide. After cooling, the content of the digestion vessel was then transferred into 50 mL volumetric flasks. The vessel was then rinsed with water and the volume of the solution was completed to 32 mL in order to achieve 3 mol L−1 HNO3 in the final solution. The solution was then used as is for injection into the FI system.Choice of the extraction resins
Most of the on-line separation systems were designed in the optic of determining a single radioelement and therefore, the choice of the extraction/ionic resin(s) used was based solely on its abilities to efficiently pre-concentrate the target analyte. For example, Epov et al.25 developed an automated system for the determination of 239,240Pu in urine designed around a flow injection analysis (FIA) system, using TRU resin. The choice of this resin was motivated by the fact that it was the most effective for Pu pre-concentration based on data published by Horwitz et al.44 However, an adverse effect of using this extraction resin is the significant co-adsorbtion of U, which is known to interfere with Pu analysis via hydride adduct and spectral overlap resulting from abundance sensitivity issue on some of the ICP-MS instrument.20 In addition, the reusability of this resin has been shown to be limited.25,27,37 For these reasons, TEVA resin was selected in this study to minimize the presence of uranium in the plutonium fraction.45,34 In addition, Am is not significantly retained onto the TEVA resin,27 facilitating the separation of 241Am and 241Pu isobars. Therefore, in order to create a multi-elemental separation, U/TEVA and n-DGA resins were added after the TEVA resin to selectively retain U and Am, respectively. The order in which the last two resins were sequentially set was based on the knowledge that U is well retained on the n-DGA43 resin while Am is not retained on UTEVA.42Calculation of detection and exposure limits
Full integration of the chromatographic peaks was used for the calculations. The relative detection limits (DL) for the individual isotopes were calculated as the concentration equivalence of three times the standard deviation of a solution produced by a blank filter (3σ, n = 5) that went through the complete procedure from ashing to column separation. In the calculation of the detection limit equivalence in air (DLEA, expressed per volume of air), the value for DL was multiplied by the digestion volume (i.e. 0.04 L) and then divided by the volume of air passed through the air filter digested (i.e. 3000 m3). Note that in case of a R/N emergency the portion of filter that would be provided for mass spectrometric determination might vary as well as the volume of air filtered. In this work we have considered that the complete filter is digested and a volume of 3000 m3 of air passed through the filters.In order to evaluate the analytical performance of the protocol developed with respect to a nuclear emergency scenario, the maximum exposure by airborne particulate (MEAP) that would result in a annual dose commitment of 0.1 mSv y−1 dose for each radioisotope was calculated. The following equation was used:

Results and discussion
Table 3, summarizes the variables tested, the optimum conditions as well as the influence of each variable on the system.| Variable | Optimal | Effect of variable on the system |
|---|---|---|
| Filter ashing | 550 °C for 4 h or 400 °C for 24 h | critical |
| Media for MW digestion | HNO3 | critical |
| Sample acidity | 3 mol L−1 HNO3 | can vary between 2–5 mol L−1 |
| Diameter of TEVA column | 4.2 mm i.d. | critical |
| Diameter of U/TEVA column | 4.2 mm i.d. | critical |
| Diameter of DGA column | 2.1 mm i.d. | critical |
| Sample flow rate | 2.5 mL min−1 | can vary between 1–5 mL min−1 |
| Eluent (U, Am) | 0.1 mol L−1 (NH4)2C2O4 | critical |
| Elution flow rate | 1 mL min−1 | critical for ICPMS performance |
| Eluent (Pu) | 0.01 mol L−1 (NH4)2C2O4 | critical |
| Effect of uranium on 239Pu determination | <10 μg L−1 uranium | critical |
| Effect of uranium on 241Am determination | <100 μg L−1 uranium | critical |
Analyte uptake conditions
The optimal conditions for the sample uptake and extraction were tested using a working solution containing the investigated actinides in 3 mol L−1 HNO3. This molarity was used since it represents the nitric concentration where the retention for Pu onto the TEVA resin is maximal.41 In the case of Am adsorption on the DGA resin and U adsorption on the U/TEVA resin, adsorptivity increases with increasing HNO3 molarity,43,49 and therefore using 3 mol L−1 HNO3 as the uptake media is adequate for sufficient retention of all the analytes onto their respective designated resin. While uranium and americium are stable in acidic conditions as U(VI) and Am(III), many oxidation states are possible for Pu, namely Pu(III), Pu(IV), Pu(V) and Pu(VI), which will have diverse distribution coefficients with selected resins. However, in 3 M HNO3, Pu(IV) is the most stable oxidation state. Therefore no attempts were made to adjust the valence of Pu.41The adsorption efficiency using different column diameters was tested using a matrix-matched standard at an uptake rate of 1 mL min−1. Two diameter sizes of columns (i.d. 2.1 and 4.2 mm) were tested for each resin. The results showed that the smaller diameter column did not contain sufficient amount of TEVA and U/TEVA resin to completely retain the analyte. For this reason, larger i.d. columns were used, which showed a complete retention of the analytes of interest. In the case of the n-DGA resin, the extraction of Am was complete with the smaller diameter column. The smaller amount of packing material required for the Am retention is probably associated with the better capacity factor and the higher selectivity achieved with the n-DGA resin.43 Therefore, for the rest of this work, 4.2 mm i.d. columns were used for TEVA and U/TEVA and a 2.1 mm i.d. column was used for DGA resin.
The effect of the sample uptake flow rate on the retention capacity of the resin was studied in order to determine the optimal flow rate for analyte retention, which also determines the time required for sample loading. This factor is critical for minimizing the overall analysis time. The maximum sample flow rate for Pu, Am, and U was tested by varying the flow rate from 1 to 5 mL min−1. No significant effect of the uptake flow rate on the adsorption of the analytes was found even for flow rates as high as 5 mL min−1. This observation is consistent with the findings from other groups using different extraction chromatography resins.25,49 While 5 mL min−1 flow rate could theoretically be used in this system, an uptake flow rate of 2.5 mL min−1 was chosen in order to minimize the time required for sample loading and rinsing steps while avoiding working at a pressure near the maximum allowable pressure of the fittings (1500 psi).
Analyte elution conditions
In order to determine the eluent that would quantitatively strip the analytes from their respective columns, three solvents were investigated: (NH4)2C2O4, HCl, at concentrations from 0.01 to 0.1 mol L−1 and H2O were tested. These solvents were chosen on the basis of their abilities to strip the analytes of interest from their respective resins.41,42,49 In addition, the ideal eluent for each analyte must produce the highest, narrow and well defined transient peaks for a better resolution between peaks for adjacent or interfering masses e.g.238U1H on 239Pu. Parameters such as peak maximum, full width half maximum (FWHM), and elution time at maximum peak were used to determine the appropriate solvent to use.It was found that 0.1 mol L−1 (NH4)2C2O4 is the most suitable solvent for the elution of Am from the n-DGA resin. The use of the cumulative recovery at maximum peak signal clearly indicates that this solvent produces a peak much narrower than H2O or HCl as shown in Fig. 2. In addition, the elution occurred much faster (∼40 s) than for the other eluents tested. The choice of the eluent for Pu from TEVA resin was not as clear as in the case of Am. The use of 0.1 mol L−1 (NH4)2C2O4 as eluent did provide the fastest elution, however, the signal intensity was not maximal, due to incomplete elution. Similarly, elution with H2O did produce a reasonable peak, but with some apparent tailing. Elution of Pu with HCl leads to a peak significantly later than the other eluents tested with no significant improvement over 0.01 mol L−1 (NH4)2C2O4 which seems to be an ideal compromise as it did provide equivalent signal and elution time than H2O, but helped minimize the effect of tailing. In the case of U adsorption onto the U/TEVA resin, 0.1 mol L−1 (NH4)2C2O4 was chosen because it provides a narrower peak with an earlier elution time. In addition, the tailing was significantly reduced using this eluent when compared to H2O. The final elution parameters used for the rest of this work are as described in Table 2.
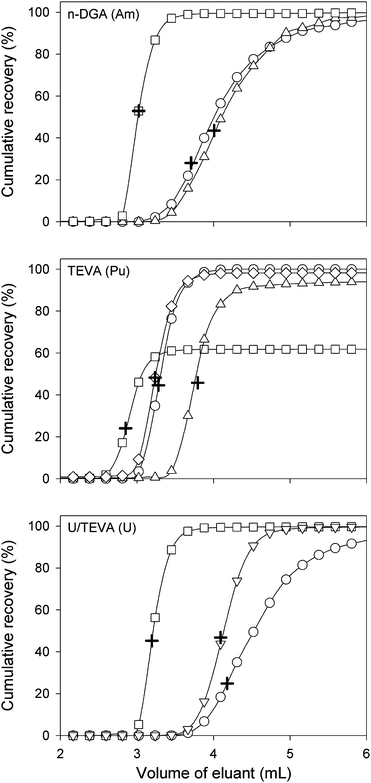 | ||
| Fig. 2 Cumulative recovery profiles for Pu, Am, and U using various elution solvents (○: H2O; ◇: 0.01 M (NH4)2C2O4; □: 0.1 M (NH4)2C2O4; ▽: 0.05 M HCl; △: 0.1 M HCl). Cumulative recovery of the radioelement at the peak maximum intensity is represented by the + symbol. | ||
Optimization of sample preparation
Emergency response to R/N event requires the development of rapid and reliable analytical procedures for fast screening of contamination. The ideal sample handling protocol must be less time-consuming, does not require experienced staff, and avoid multistage procedures that would complicate the analytical routines and increase risks of contamination.Many methods have been published on the preparation of atmospheric particulate samples for chemical analysis based on digestion and extraction. However, airborne particulate matter collected on membrane filters is a difficult to digest sample because it contains a variety of matrix constituents, such as organics, oxides and silicates and therefore acidic decomposition is a critical step and in some cases total digestion is required particularly when a small amount of particles is collected on the filters.
In this work, for the in-house filters (PP), we have investigated whether it was necessary to completely dissolve the filters for effective extraction of the radioisotopes and whether it was possible to omit the dry ashing step to speed-up the process of sample preparation. The direct digestion of the whole filters in the microwave, without the dry ashing step, created a high pressure and could cause minor explosions inside the MW cavity. The leaching of PP filters in concentrated HNO3 caused the filter to aggregate and float on the surface of the acid, leading us to believe that efficiency of the extraction would be limited. Therefore, the ashing of the filters followed by digestion with HNO3 in a MW was found the most suitable procedure and produced nearly complete dissolution of the filters. Since time is a crucial issue in emergency situations, the effect of temperature and time of ashing step was also investigated. We have compared dry ashing the filters at 400 °C for 24 h to 550 °C for 4 h. It was found that recoveries obtained for all the radioisotopes investigated were comparable. Therefore, in case of emergencies, the short procedure is recommended.
The digestion of urban particulate matter (SRM-1648) resulted in a slight precipitate probably due to the incomplete dissolution of silica in HNO3 only and this will be probably the case of real loaded filters. Therefore the addition of HF during the digestion was investigated. It was found that the use of HF produced clear digest solutions and the recoveries for uranium and americium were comparable to the procedure using HNO3 only. However Pu isotopes suffered from low recoveries, 10 to 20%, in the spiked samples and therefore for the rest of this work, the use of HF was omitted. The samples were filtered before column separation to avoid clogging of the columns. Similar low recoveries for 242Pu were observed by Komosa et al.50 in their work when they compared different acid combinations, including HNO3/HF/HClO4 for leaching of Pu from air filters made of chlorinated polyvinyl chloride.
It is important to note that the efficiency of the sample preparation optimized in this work was evaluated only for our analytes of interest. An additional advantage of this sample preparation protocol is that the resulting acidity of the digested samples is optimal for loading onto the separation columns, with no additional pre-treatment.
Analytical figures of merits
The analytical precision of this automated system was evaluated at two concentration levels for Pu, Am, and U. The precision level varies from 0.89% to 5.66% for five replicates (Table 4). The choice of suitable eluent for each element and the quantitative elution resulted in sharp and well defined chromatographic peaks (Fig. 3) which have contributed to the high precision when using peak area integration. The level of precision achieved using the automated system is adequate for case of emergency response, as a precision level below 10% would most-likely be deemed sufficient51 as well as for environmental monitoring. Table 3 shows the detection limits (DL) achievable for U, Pu and Am isotopes using the sequential separation protocol in solution, using only 5 mL sample, and in air (based on 3000 m3 of air). The DLs were estimated from successive replicates of blank filters and found to be 0.0006 (238U), 0.0063 (239Pu), 0.0041 (240Pu), 0.062 (241Am) μBq m−3 based on 3000 m3 of air filtered.| Sample volume, mL | 5 | |||
| Time of analysis, min | 16 | |||
| Sample frequency, sample d−1 | 80 | |||
| Sensitivity (105 counts pg−1) | 1.69 (242Pu), 1.81 (243Am), 1.64 (238U) | |||
| Precision, % RSD (5 replicates) | 50 pg L−1 | 150 pg L−1 | 1 μg L−1 | 2 μg L−1 |
| 242Pu | 3.97 | 3.50 | — | — |
| 243Am | 5.66 | 4.53 | — | — |
| 238U | — | — | 0.89 | 2.86 |
 | ||
| Fig. 3 Chromatogram of the sequential separation for PP filters spiked with 5 μg L−1 (0.06 Bq L−1) U, 0.14 Bq L−1242Pu and 1 Bq L−1243Am. | ||
The analytical performances of the present work were assessed against exposure risk to radionuclides from airborne particulate as shown in Table 5. As can be seen from column 5 and 6, the limits of quantifications (evaluated as 5 times the DL) for all radioisotopes investigated are several orders of magnitude below the maximum exposure limit by airborne particulate which makes this technique particularly suitable for both emergency purposes and for routine radiological monitoring of air.
| Isotope | Half-life (y) | Effective dose conversion factor (Sv Bq−1)46,47 | Detection limits (mBq L−1) | Detection limit equivalence in air (DLEA, mBq m−3) | Maximum exposure by airborne particulate (MEAP, mBq m−3) |
|---|---|---|---|---|---|
| 238U | 4.46 × 109 | 3.2 × 10−5 | 0.052 | 5.60 × 10−7 | 0.386 |
| 239Pu | 2.411 × 104 | 8.1 × 10−5 | 0.595 | 6.34 × 10−6 | 0.152 |
| 240Pu | 6.537 × 103 | 8.1 × 10−5 | 0.392 | 4.18 × 10−6 | 0.152 |
| 242Pu | 3.76 × 105 | 7.92 × 10−5 | 0.0022 | 2.34 × 10−8 | 0.156 |
| 241Am | 432.2 | 1.2 × 10−4 | 5.80 | 6.1 × 10−5 | 0.103 |
| 243Am | 7.37 × 103 | 1.19 × 10−4 | 0.088 | 9.38 × 10−7 | 0.104 |
A significant challenge when quantifying Pu and Am by ICP-MS is the presence of U at high concentrations. In this work we have investigated the effect of increasing levels of uranium in the sample on m/z= 239, 240, 241, 242 and 243 to determine the maximum concentration of uranium at which the separation protocol became obsolete for Pu and Am determination. As shown in Fig. 4, the presence of U will affect the background at masses from 239 and above, depending on the concentration of uranium present in the sample. In this work we determined that the ratio of the background signal at masses 239, 240 and 241 to the signal of uranium represent only 3 × 10−7, 4 × 10−8 and 3 × 10−8 for 239Pu, 240Pu and 241Am, respectively. However, the presence of uranium at a level of 10 μg L−1 in the loading sample is shown to be sufficient to prevent proper identification of 10 mBq L−1239Pu, which represents 2 times the quantification limit achieved with this method. The same level of uranium (10 μg L−1) was found not to affect the quantification limits for 240Pu and 241Am. However, the quantification limits for both 240Pu and 241Am decreases by a factor of 10 in the presence of 100 μg L−1 of uranium in the measured solution. As a result, it appears that the present method is suitable for the simultaneous quantification of 238U, 239Pu, 240Pu and 241Am in air for environmental monitoring purposes since uranium levels in air are unlikely to be higher than the maximum tolerated levels of uranium to allow the simultaneous quantification of radionuclides. However, the application of this method to a radiological event monitoring should be taken with caution, considering that presumably the uranium concentration will be significantly higher than the tolerable maximum level identified for the present method.
 | ||
| Fig. 4 Effect of increasing levels of uranium in loading sample on the determination of (A) 239Pu, (B) 240Pu, (C) 242Pu and (D) 241Am. | ||
Contrary to the other automated systems that were developed in the past for the measurement of actinides,25,29,35,37 this system is designed in such a way that the resins can be reused multiple times. This helps improving the sample throughput and decreases the analytical cost per sample by reducing the amount of resins used.27 However, this situation could lead to possible sample cross-contamination resulting from sample carry-over. Therefore, in order to assess the level of cross-contamination, a sequence composed of 1 spiked sample (1 Bq L−1 Pu and Am; 2 μg L−1 U) followed by 3 blank solutions (3 mol L−1 HNO3) was used. The amount of each actinide was reported as a percentage of the signal compared to the one obtained by the sample. Carry-over percentages of 0.32, 0.73, and 0.02% for U, Pu, and Am, respectively, were measured in the first blank solution. Undetectable carry-over levels were found for the second rinse. These levels of carry-over are comparable to those found by others using similar extraction chromatography resins.29,34,51 Considering that the maximum carry-over rate is below 1% for all analytes, this indicates that a range of 100-fold in the analyte concentration will not significantly affect the quantification process. Therefore, in case of emergency, only a consecutive sample that exhibits a concentration difference of 100-fold should be retreated after either re-rinsing the resins or changing the columns, depending on the time and resources available.
Measurements of actinides in spiked samples
In this work, the effectiveness of separation/detection method was determined by spiking PP blank filters, SRM-1648 particulate matter and SRM-2783 filter with a concentration of radionuclides 5-times lower than the maximum exposure levels in airborne particulate. For this work, we have considered for all isotopes the same maximum exposure of 0.10 mBq m−3 corresponding to 63 mBq per sample (or 2.5 Bq L−1 in 25 mL of digested samples). Uranium was spiked in PP filters at a level of 5 μg L−1.The levels of uranium in the reference materials and the recoveries of spiked radioisotopes are compiled in Table 6. The concentration of uranium found in the SRM-2783 loaded filter with particulate matter agrees well with the observed values reported by Kulkarni52 and NIST. These results also show the capability of this method for accurate determinations of uranium at low ng L−1 levels which makes this method particularly attractive for monitoring of uranium levels in air. The concentration of uranium reported for the urban particulate matter (SRM-1648) was calculated with a 2.7% (RSD) precision and a bias of 6% from the certified value which demonstrates the efficiency of sample preparation methodology optimized in this work. This value, however, lay well outside the uncertainty interval of the certified value which could be explained by the fact that this reference material was purchased many years ago and the content of some elements is possibly altered. Note that the urban particulate matter SRM-1648 has been replaced by SRM-1648a which is the same particulate matter than SRM-1648 that has been re-blended and reanalyzed. Curiously, no value for uranium is provided for SRM-1648a. Another possible explanation to the lower recoveries of uranium in the urban dust is the acid resistant residual material, for example, refractory oxide that requires the use of more drastic sample preparation conditions such as fusion.
| Sample | U, mg Kg−1 | U, μg L−1 | 239Pu, Bq L−1 | 240Pu, Bq L−1 | 241Am, Bq L−1 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Found | Certified | Added | Found | Added | Found | Added | Found | Added | Found | |
| a Only reference value is provided by NIST. b Reference value from Ref. 52. | ||||||||||
| SRM-2783 Air particulate matter on filter | 1.216 ± 0.054 | 1.234 ± 0.024a 1.2 ± 0.1b | — | — | 2.52 | 2.44 ± 0.15 | 2.52 | 2.42 ± 0.02 | 2.52 | 2.36 ± 0.18 |
| SRM-1648 Urban dust | 5.17 ± 0.14 | 5.5 ± 0.1 | — | — | 2.52 | 2.34 ± 0.13 | 2.52 | 2.29 ± 0.20 | 2.52 | 2.32 ± 0.20 |
| Spiked PP filter | — | — | 5 | 4.66 ± 0.23 | 2.52 | 2.31 ± 0.21 | 2.52 | 2.30 ± 0.15 | 2.52 | 2.44 ± 0.15 |
PP filters are being used by Health Canada in various stations throughout Canada for air sampling for the monitoring of radioactivity in air. Therefore it was important for us to investigate the efficiency of this technique for the multi-isotopic determination of U, Pu and Am in air samples in a single filter (or a portion of a filter). As shown in Table 5, the recoveries for U, Pu and Am in the spiked PP filters were in the range 90 to 95%.
Conclusion
The sequential and automated determination of U, Pu and Am isotopes in a single sample by ICP-MS makes the present method suitable for both emergency screening and routine environmental monitoring of radioactivity. The total sample analysis is significantly reduced compared to conventional radiometric methods and offers advantages such as automated separation, on-line coupling of the separation and detection, less cross-contamination and less risk of exposure of the operator to chemicals and reduced analytical errors associated to the analysts. The sensitivity and the precision of this method guarantees its suitability for routine environmental monitoring for the determination of low levels of actinides in air.Acknowledgements
The authors would like to thank the McMaster Co-op students Kyle Verdecchia, Rachel Timmins, Amanda Fawcett and Vera Kochermin that were involved at various stages of development and testing of the system.References
- E. Hrnecek, P. Steier and A. Wallner, Appl. Radiat. Isot., 2005, 63, 633–638 CrossRef CAS.
- O. B. Egorov, M. J. O'Hara, R. S. Addleman and J. W. Grate, In Radioanalytical Methods in Interdisciplinary Research; ed. C. A. Laue and K. L. Nash, 2004; Vol. 868, pp 246–270 Search PubMed.
- C. S. Kim, C. K. Kim and K. J. Lee, J. Anal. At. Spectrom., 2004, 19, 743–750 RSC.
- E. R. Gonzales, S. R Garcia, C. Mahan and W. J. Hang, J. Radioanal. Nucl. Chem., 2005, 263, 457–465 CrossRef CAS.
- D. L. Stricklin, A. Tjarnhage and U. J. Nygren, J. Radioanal. Nucl. Chem., 2002, 251, 69–74 CrossRef CAS.
- I. Friberg, Appl. Radiat. Isot., 1999, 50, 365–373 CrossRef CAS.
- N. Green, Sci. Total Environ., 1993, 130, 207–218 CrossRef.
- V. P. Reshetin, Atmos. Environ., 2005, 39, 4471–4477 CrossRef CAS.
- K. G. Andersson, C. L. Fogh, M. A Byrne, J. Roed, A. J. H. Goddard and S. A. M. Hotchkiss, Health Phys., 2002, 82, 226–232 CrossRef CAS.
- V. A. Kutkov, Radiat. Prot. Dosim., 2000, 89, 263–266 CAS.
- R. Pollanen and T. Siiskonen, J. Environ. Radioact., 2006, 87, 279–288 CrossRef CAS.
- R. Pollanen and T. Siiskonen, Health Phys., 2006, 90, 167–175 CrossRef CAS.
- D. E. Farmer, A. C. Steed, J. Sobus, K. Stetzenbach, K. Lindley and V. F. Hodge, Health Phys., 2003, 85, 457–465 CrossRef CAS.
- Z. Holgye and R. Filgas, Health Phys., 2006, 90, 328–336 CrossRef CAS.
- A. A. Marchetti, T. A. Brown, C. C. Cox, T. F. Hamilton and R. E. Martinelli, J. Radioanal. Nucl. Chem., 2005, 263, 483–487 CrossRef CAS.
- C. Vockenhuber, I. Ahmad, R. Golser., W. Kutschera, V. Liechtenstein, A. Priller, P. Steier and S. Winkler, Int. J. Mass Spectrom., 2003, 223, 713–732 Search PubMed.
- J. E. Mc Aninch and T. F. Hamilton, Measurements of plutonium and other actinides elements at The Center for Accelerator Mass Spectrometry: Comparative assessment of competing techniques; Lawrence Livermore National Laboratory, Livermore CA, UCRL-ID-133118, 1999 Search PubMed.
- N. L. Elliot, G. A. Bickel, S. H. Linauskas and L. M. Paterson, J. Radioanal. Nucl. Chem., 2006, 267, 637–650 CrossRef CAS.
- S. P. LaMont, C. R. Shick, P. Cable-Dunlap, D. J. Fauth and T. R. LaBone, J. Radioanal. Nucl. Chem., 2005, 263, 477–481 CrossRef CAS.
- D. Lariviere, V. F. Taylor, R. D. Evans and R. J. Cornett, Spectrochim. Acta, Part B, 2006, 61, 877–904 CrossRef.
- J. S. Becker, Spectrochim. Acta, Part B, 2003, 58, 1757–1784 CrossRef.
- J. S. Becker and H. J. Dietze, Int. J. Mass Spectrom., 2000, 197, 1–35 Search PubMed.
- G. Lujaniene, J. Šapolaite, V. Remeikis, V. Lujanas and A. Jermolajev, Czech. J. Phys., 2006, 56, D55–D61 CAS.
- K. Benkhedda, V. N. Epov and R. D. Evans, Anal. Bioanal. Chem., 2005, 381, 1596–1603 CrossRef CAS.
- V. N. Epov, K. Benkhedda, R. J. Cornett and R. D. Evans, J. Anal. At. Spectrom., 2005, 20, 424–430 RSC.
- D. Schaumloffel, P. Giusti, M. V. Zoriy, C. Pickhardt, J. Szpunar, R. Lobinski and J. S. Becker, J. Anal. At. Spectrom., 2005, 20, 17–21 RSC.
- W. Hang, L. W. Zhu, W. W. Zhong and C. Mahan, J. Anal. At. Spectrom., 2004, 19, 966–972 RSC.
- T. Seki and K. Oguma, Bunseki Kagaku, 2004, 53, 353–357 CrossRef CAS.
- C. S. Kim and C. K. Kim, Anal. Chem., 2002, 74, 3824–3832 CrossRef CAS.
- J. H. Aldstadt, J. M. Kuo, L. L. Smith and M. D. Erickson, Anal. Chim. Acta, 1996, 319, 135–143 CrossRef CAS.
- Y. Ohtsuka, Y. Takaku, K. Nishimura, J. Kimura, S. Hisamatsu and J. Inaba, Anal. Sci., 2006, 22, 309–311 CrossRef CAS.
- Y. Ohtsuka, Y. Takaku, J. Kimura, S. Hisamatsu and J. Inaba, Anal. Sci., 2005, 21, 205–208 CrossRef CAS.
- J. B. Truscott, P. Jones, B. E. Fairman and E. H. Evans, J. Chromatogr., A, 2001, 928, 91–98 CrossRef CAS.
- C. S. Kim, C. K. Kim, J. I. Lee and K. J. Lee, J. Anal. At. Spectrom., 2000, 15, 247–255 RSC.
- V. N. Epov, K. Benkhedda and R. D. Evans, J. Anal. At. Spectrom., 2005, 20, 990–992 RSC.
- P. Evans, S. Elahi, K. Lee and B. Fairman, J. Environ. Monit., 2003, 5, 175–179 RSC.
- O. B. Egorov, M. J. O'Hara, O. T. Farmer and J. W. Grate, Analyst, 2001, 126, 1594–1601 RSC.
- D. Solatie, P. Carbol, M. Betti, F. Bocci, T. Hiernaut, V. V. Rondinella and J. Cobos, Fresenius J. Anal. Chem., 2000, 368, 88–94 CrossRef CAS.
- M. J. Betti, J. Chromatogr., A, 1997, 789, 369–379 CrossRef CAS.
- M. R. Smith, O. T. Farmer, J. H. Reeves and D. W. Koppenaal, J. Radioanal. Nucl. Chem., 1995, 194, 7–13 CAS.
- E. P. Horwitz, M. L. Dietz, R. Chiarizia, H. Diamond, S. L. Maxwell and M. R. Nelson, Anal. Chim. Acta, 1995, 310, 63–78 CrossRef CAS.
- E. P. Horwitz, M. L. Dietz, R. Chiarizia, H. Diamond, A. M. Essling and D. Graczyk, Anal. Chim. Acta, 1992, 266, 25–37 CrossRef CAS.
- E. P. Horwitz, D. R. McAlister, A. H. Bond and R. E. Barrans, Solvent Extr. Ion Exch., 2005, 23, 319–344 CrossRef CAS.
- E. P. Horwitz, R. Chiarizia, M. L. Dietz and H. Diamond, Anal. Chim. Acta, 1993, 281, 361–372 CrossRef CAS.
- M. V. Zoriy, P. Ostapczuk, L. Halicz, R. Hille and J. S. Becker, Int. J. Mass Spectrom., 2005, 242, 203–209 Search PubMed.
- K. F. Eckerman, A. B. Wolbarst and A. C. B. Richardson, Limiting Values of Radionuclide Intake and Air Concentration and Dose Conversion Factors for Inhalation, Submersion, and Ingestion; U.S. Environmental Protection Agency, Washington, D.C., EPA-5201/1-88–020, 1988 Search PubMed.
- International Commission on Radiological Protection, Annals of the ICRP 1988, 19, 1–23 Search PubMed.
- International Commission on Radiological Protection, Annals of the ICRP 1995b, ICRP Publication 71. Ann. ICRP 25(3/4) Search PubMed.
- J. W. Grate and O. B. Egorov, Anal. Chem., 1998, 70, 3920–3929 CrossRef CAS.
- A. Komosa, S. Chibowski and J. Orzet, Nukleonika, 2001, 46, 151–153 Search PubMed.
- V. Bolyatko, N. Goleminov, A. Zvantsev, E. Kramer-Ageev, N. Mogilenets and V. Troshin, Radiat. Prot. Dosim., 2005, 116, 148–151 CrossRef CAS.
- P. Kulkarni, S. Chellam, J. B. Flanagan and R. K. M. Jayanty, Anal. Chim. Acta, 2007, 599, 170–176 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |