Quantitative assay of urinary hepcidin using MALDI-TOF mass spectrometry
Melvin C. L.
Gay
ab,
Ian
Mullaney
b,
Debbie
Trinder
cd,
John K.
Olynyk
cde and
Robert D.
Trengove
*a
aSeparation Science & Metabolomics Laboratory, Murdoch University, South Street, Murdoch, 6150, Western Australia, Australia. E-mail: R.Trengove@murdoch.edu.au
bSchool of Pharmacy, Murdoch University, Western Australia, Australia
cSchool of Medicine and Pharmacology, University of Western Australia, Fremantle Hospital, Fremantle, Western Australia, Australia
dWestern Australian Institute for Medical Research, Nedlands, Western Australia, Australia
eThe Department of Gastroenterology, Fremantle Hospital, Fremantle, Western Australia, Australia
First published on 22nd December 2009
Abstract
Hepcidin has been identified as the principle regulatory hormone essential for iron homeostasis. Quantitative analysis of hepcidin in bodily fluids provides an insight into the pathogenesis of disorders of iron metabolism such as hereditary hemochromatosis and anemia of chronic disease. This study describes the use of solid phase extraction (SPE) as a preparative step followed by matrix assisted laser desorption/ionization-orthogonal-time-of-flight mass spectrometry (MALDI-TOF MS) with internal standard for the quantitative analysis of unlabelled urinary hepcidin. More than 70% extraction recovery of hepcidin (hepcidin-25) with monoisotopic resolution was achieved. Urinary creatinine was analyzed using HPLC-UV/Vis with hepcidin-25 levels of 2.2 to 2.7 nmol/mmol of creatinine observed in healthy controls. Spot-to-spot variation of hepcidin standard additions was less than 3.5%. Intra- and inter-day precision assay of less than 9.5% relative standard deviation was achieved with less than 0.5% variation between the intra-day assay data. In summary, a validated non-invasive method has been developed for the quantification of unlabelled urinary hepcidin-25 which can be used to screen for iron-related disorders such as hemochromatosis in iron-overloaded patients.
Introduction
Hepcidin is a cysteine-rich antimicrobial peptide that was first isolated from human urine1 and human plasma ultrafiltrate.2 Hepcidin is produced by hepatocytes and exists in 3 forms that differ by amino-terminal truncation. It is then secreted into plasma and excreted in urine.1,2 Preprohepcidin, which contains 84 amino acids, is synthesized in the liver and cleaved by a signal peptidase to prohepcidin (60 amino acids). It is subsequently cleaved by the prohormone convertase furin3 to the shorter forms of hepcidin (Hepc-20, -22 and -25), with Hepc-20 and -25 being the predominate forms. These contain 20 and 25 amino acid residues, respectively, with all 8 cysteines connected by intramolecular disulfide bonds.Bioactive Hepc-25 is a circulating peptide that regulates systemic iron homeostasis, coordinating the gastrointestinal absorption, macrophage recycling and storage of iron.4 Hepcidin is a negative regulator of iron absorption and iron release from macrophages. It binds to the cellular iron export protein ferroportin which is highly expressed on the basolateral surface of the intestine and macrophages,5 causing ferroportin internalization and degradation. The loss of ferroportin from the cell surface prevents cellular iron export resulting in decreased iron absorption and impaired iron release from macrophages. Conversely, decreased expression of hepcidin leads to increased iron absorption and release of iron from macrophages due to the increased levels of cell surface ferroportin.6
Hepcidin synthesis is induced by iron overload7 and inflammation,8 primarily through the actions of inflammatory cytokines such as interleukin-6 (IL-6).9,10 Increased levels of urinary hepcidin have been detected in patients with hypoferremia due to sepsis or anemia of chronic disease, both manifesting features of iron restricted erythropoiesis.9,11
Hepcidin deficiency has been linked to hypoxia,8 iron deficiency,7 and anemia,8 especially those associated with iron-loading anemias as a result of ineffective erythropoiesis and increased intestinal iron absorption (for example, β-thalassemia).12 Hypoxia stimulates hepcidin by stabilizing hypoxia-inducible factor-1 which inhibits hepcidin transcription.13 Hypoxia also stimulates erythropoietin production which in addition to its effects on erythropoiesis, may also directly down-regulate hepcidin antimicrobial peptide expression in hepatocytes by a mechanism involving the transcription factor C/EBPα.14
Impaired hepcidin expression has also been shown in the disorder hereditary hemochromatosis, which results from mutations in the key iron-regulatory genes HFE, hemojuvelin (HJV), transferrin receptor 2 (TFR2) and hepcidin antimicrobial peptide (HAMP). These mutations cause excessive intestinal iron absorption from the diet, leading to the iron overload.12,15–17
As a result of impaired hepcidin regulation in hereditary hemochromatosis, detection and quantification of hepcidin levels would substantially facilitate diagnosis and characterization of the likely aetiology, especially when combined with serum iron parameters and hepatic iron concentration. Several methods have been developed for both the detection and quantification of hepcidin. For example, Ganz et al.18 have developed an immunodot assay for both serum and urinary hepcidin with limit of detection at 5 ng/mL while Kulaksiz et al.19 developed an ELISA detection assay for pro-hepcidin.
There have been many studies demonstrating the usefulness of time-of-flight mass spectrometry based techniques for targeted peptide quantification.20–23 In particular, the level of endogenous hepcidin was quantified using surface-enhanced laser desorption/ionization time-of-flight mass spectrometry (SELDI-TOF MS) in several studies.24–28 Both Kemna et al.24 and Swinkels et al.25 have employed a direct on-spot method using a copper-loaded immobilized metal-affinity capture ProteinChip array (IMAC30-Cu2+) for the quantitation of hepcidin. Swinkels et al.25 included an internal standard (hepcidin-24) into the assay. Tomosugi and co-workers26 used serum/urine with a bioprocessor and the level of Hepc-25 was analyzed using external standards. The method by Swinkels and co-workers25 requires 495 µL of urine and is difficult to automate due to the on-spot procedures. The assay by Altramure et al.27 which is similar to Tomosugi et al.26 assay, semi-quantitated urinary hepcidin using external references containing ACTH, bovine insulin and synthetic human hepcidin. The hepcidin concentration was then normalized to creatinine and urea level in the body. Ward et al.28 utilized a stable isotope labeled hepcidin internal standard in the SELDI assay. Bansal et al.29 had employed weak cation-exchange (WCX) magnetic nanoparticles and MALDI-TOF MS for the quantification of urinary hepcidin using [15N,13C2]Gly12,20-hepcidin as internal standard.
Three serum hepcidin quantitative methods using liquid chromatography tandem triple quadrupole mass spectrometry (LC-MS/MS) together with solid phase extraction (SPE) were published with Murphy et al.30 using 100 µL aliquot of plasma or serum and calcitonin gene-related peptide (CGRP) as internal standard. The limit of quantification (LOQ) for Murphy et al.30 assay was 0.36 pmol hepcidin/mL of serum. Kobold et al.31 employed isotope-dilution on 45 µL of serum and µHPLC-tandem MS for hepcidin analysis with a LOQ of 0.3 pmol/mL of serum and Li et al.32 used [13C9,15N1-Phe4]-hepcidin as internal standard for the analysis of serum samples with a LOQ of 0.90 pmol/mL. Bansal et al.33 reported the first LC-tandem ion trap MS for the quantitation of urinary hepcidin using selected reaction monitoring (SRM) of 930 m/z with a LOQ of 1 pmole/mL. Murao et al.34 employed peptide precipitation method using trichloroacetic acid and quantification of hepcidin with LC-MS/MS.
In summary, several authors had utilized different external calibration standards,26,27 stable isotope labeled hepcidin28,29,31–33 or truncated hepcidin analogue25,35 as internal standards for the quantification of hepcidin. The use of external standards are only able to provide semi-quantitative hepcidin concentrations whereas the use of labeled hepcidin are costly and difficult to synthesize. The quantification of endogenous hepcidin using unlabeled hepcidin will be ideal in providing an accurate and cost effective assay.
The advantage of using MALDI-TOF MS based assays mentioned here is the potential to simultaneously distinguish and quantify multiple isoforms/variants of a particular protein or peptide when compared to most ELISA based assays.24–29 LC-MS based assays required about 10 min for each analysis whilst the MALDI-TOF assay requires less than 1 min of analysis time.29 However, both techniques are complementary to each other where LC-MS can be used to provide further structural confidence for hepcidin while MALDI-TOF MS is able to provide fast and accurate analysis.
In this study, we employ the use of micro solid phase extraction tips (µ-SPE) which are easy to operate and requires only microliter amounts of biological fluid without any pretreatment. We combined this with a high resolution matrix assisted laser desorption/ionization-orthogonal-time-of-flight mass spectrometer (MALDI-TOF MS) which is able to provide sensitive analysis to low femtomole levels and have developed a high-throughput assay that is suitable for the quantification of urinary endogenous hepcidin and for biomarker discovery. This is the first reported assay which employs standard addition method for the quantification of endogenous hepcidin.
Experimental
Materials
Trifluoroacetic acid (TFA) was obtained from Sigma-Aldrich (St. Louis, MO). HPLC-grade methanol (MeOH) and acetonitrile (ACN) were obtained from Burdick and Jackson Inc. (Muskegon, MI). MilliQ water was obtained from an in-lab Milli-Q system. Mid-stream urine samples from healthy subjects were collected in sterile centrifuge tubes and immediately divided into 150 µL aliquots and stored in microcentrifuge tubes at −80 °C. The study was carried out in accordance with the protocols approved by the Fremantle Hospital Human Research Ethics Committee, and all subjects gave informed consent.Extraction of urinary hepcidin
The Omix C-4 tips (Varian Inc.) were conditioned with 50% ACN/H2O (v/v) followed by 1% TFA/H2O (v/v). 100 µL of urine sample without any pre-treatment was drawn into the Omix tip. The tip was then washed twice with 30% MeOH/H2O (v/v) buffered to pH 10 with ammonium hydroxide (Sigma-Aldrich, MO),32 and eluted with 20 µL 80% ACN/H2O/1% TFA (v/v/v). All eluents were transferred into 600 µL microcentrifuge tubes for further analysis.Preparation of hepcidin standard and internal standard (IS)
Synthetic hepcidin standard (DTHFPICIFCCGCCHRSKCGMCCKT, 2789.4 g/mol) was obtained from Peptide Institute Inc. (Osaka, Japan). Stock solutions of 2 µM hepcidin was diluted with 50% ACN/H2O (v/v) to make final concentrations of 20 nM, 50 nM, 200 nM or 500 nM.Adrenocorticotropic hormone (ACTH) fragment 18–39 (RPVKVYPNGAEDESAEAFPLEF, 2464.2 g/mol) was obtained from Sigma-Aldrich (St. Louis, MO) and was used as internal standard (IS). Stock solution of 10 µM ACTH was diluted to 100 nM with 50% ACN/H2O (v/v).
One part of the internal standard ACTH was mixed with one part of 20 nM, 50 nM, 200 nM or 500 nM hepcidin concentration to give a final concentration of 50 nM ACTH and hepcidin standard at 10 nM, 25 nM, 100 nM and 250 nM respectively. For ‘blank’ samples, one part of ACTH was mixed with one part of 50% ACN/H2O (v/v).
Matrix-assisted laser desorption/ionization orthogonal time-of-flight MS (MALDI-TOF MS)
MALDI matrix (α-cyano-4-hydroxycinnamic acid, CHCA) was obtained from Sigma-Aldrich (St. Louis, MO). The matrix was re-crystallized using HPLC/Spectrophotometric grade ethanol (Sigma-Aldrich, MO). Ten mg/mL (w/v) of CHCA matrix was dissolved in a solution consisting of 0.05 mM pyridine (UNIVAR, NSW) in MeOH/ACN/H2O (36 : 56 : 8; v/v/v) with 1% TFA.36,37A standard addition and dried droplet method was used for the spiking and spotting of the extracted sample. One part of extracted sample was spiked with one part of the mixture (containing internal standard and specific concentration of hepcidin or ‘blank’ sample) and two parts of CHCA matrix.
A 0.8 µL aliquot of the mixture was spotted onto a disposable 384-well MALDIchip (PerkinElmer) and allowed to air dry. Five replicates of each concentration were spotted onto the MALDIchip.
Analysis was performed using a MALDI-TOF mass spectrometry (prOTOF™ 2000, PerkinElmer). A 9-point circle pattern for automated sample acquisition was used in which 10 positions on the MALDI spot were sampled and 100 shots per spot were acquired twice. A total of 2000 shots were averaged to yield a representative spectrum. The MALDI-TOF was operated in positive ion reflector mode with laser rate and energy set at 100.0 Hz and 70%. The declustering voltage was set at 35 V and the acceleration voltage was set at 16 kV. Nitrogen cooling and focusing flow was set at 190.0 mL/min and 212.1 mL/min respectively. Data were collected between 1000 and 5000 Da. External mass calibration was performed using a ProXPRESSION MALDI calibration kit (PerkinElmer, MA). Data acquisition was performed using TOFworks and analyzed using Data Explorer v4.0 software (Applied Biosystems) and Progenesis PG600 (Nonlinear Dynamics).
Statistics and method validation
Calibration curves were constructed from the summed peak area ratio of the analyte (2788 to 2794 m/z) to the internal standard (2465 to 2469 m/z) versus theoretical concentrations. Standard curves were plotted and a linear equation (y = mx + b) derived where y is the relative area (analyte/IS) and x is the standard addition concentration of analyte. Unknown endogenous sample concentrations were calculated using the equation x = −(−b/m).Spot-to-spot reproducibility, intra and inter-day precision assay
Spot-to-spot reproducibility was determined by replicate spotting (n = 5) of extracted urine samples with different concentrations of added hepcidin together with internal standard and compared with different sets (n = 15).Intra- and inter day reproducibility was determined using a single urine sample that had been aliquoted into smaller tubes and was freeze-thawed only once. For the intra-day precision study, a set of 5 replicates was performed using the hepcidin extraction method mentioned above. This was repeated on 3 separate days for the assessment of inter-day precision.
All precision measurements (coefficient of variation) were expressed as the percentage relative standard deviation (%RSD).
Stability of hepcidin-25 at 30 °C and the influence of multiple freeze-thaw cycles
For stability testing, 1 mL of urine aliquots from the same urine sample were incubated in a thermomixer (Eppendorf) at 30 °C with moderate vortex of 500 RPM. The samples were analyzed at time 0, 1, 3, 5, 7, 24, 48 and 72 hours using the hepcidin extraction method mentioned above.One milliliter aliquots from the same urine sample were stored at −80 °C and underwent multiple freeze-thaw cycles. Samples were thawed in a thermomixer (Eppendorf) at 30 °C with vortex of 500 RPM for an hour. A maximum of 4 freeze-thaw cycles were investigated.
Creatinine analysis
Creatinine standard (Sigma-Aldrich, MO) was diluted to a concentration range of 15 to 345 µg/mL with 10.8 mM ammonium acetate solution (Univar, NSW). Urine samples were diluted 10 times with 10.8 mM ammonium acetate solution prior to analysis. Standards and samples were injected (1 µL) into an Agilent 1100 HPLC with a diode array detector (Agilent Technologies) with a 150 mm × 2.0 mm × 3 µm Pursuit XRs diphenyl column (Varian Inc.). The column was maintained at 25 °C with a flow rate of 0.2 mL/min. Mobile phase A consisted of 10 mM ammonium acetate solution and mobile phase B was ACN with 0.1% formic acid (v/v), a gradient program holding at 0% B for 10 min followed by ramping to 100% B at 20% B/min was used. The eluant was monitored at a wavelength of 236 nm.Results and discussion
Extraction of hepcidin
The MALDI-TOF mass spectrum of extracted urine sample agreed with the observations of Park et al.1 and Kemna et al.,35 showing 2 pre-dominant forms of hepcidin (Hepc-20 and Hepc-25) with the monoisotopic masses of 2190 and 2788 m/z respectively (Fig. 1). The Hepc-22 with monoisotopic mass of 2434 m/z was present at very low levels. Monoisotopic resolution of hepcidin was not observed in SELDI-TOF MS analysis24–27,35 as most SELDI-TOF instruments can only perform measurements in linear mode and produces low resolution mass spectra.29 The orthogonal ion extraction design coupled with the reflectron capabilities of the MALDI-TOF MS, on the other hand, is able to provide improved mass accuracy,38,39 thus demonstrating the presence of monoisotopic resolutions for Hepc-20, Hepc-25, ACTH and even the low level compound like Hepc-22.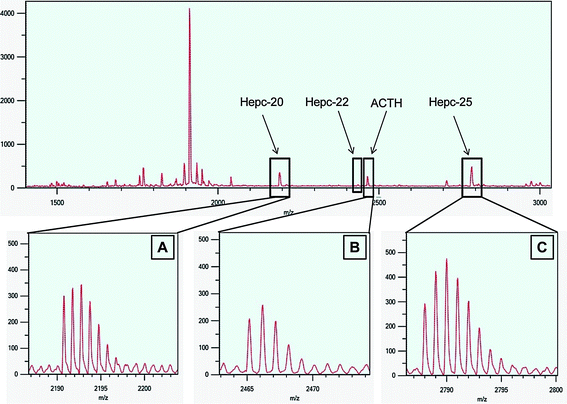 | ||
| Fig. 1 Extracted urine profile using MALDI-TOF-MS. MALDI-TOF mass spectra of extracted urine sample over the mass range 1400–3000 m/z. Urinary hepcidin-20 and -25 are shown with monoisotopic masses of 2191 (A) and 2788 (C) m/z respectively. Internal standard (ACTH) has a monoisotopic mass of 2465 m/z (B) and the position of hepcidin-22 with monoisotopic mass of 2435 m/z. | ||
The use of internal standard minimized variability in analyte signal intensities and improved experimental reproducibility. Adrenocorticotropic hormone (fragment 18–39), which has a monoisotopic mass of 2465 m/z, was selected as an internal standard as it was readily available, inexpensive and is not present in urine samples. The ACTH peak is situated between Hepc-20 and -25 and it does not overlap with Hepc-22 which makes ACTH an ideal internal standard for hepcidin analysis. The use of stable isotope labeled hepcidin or hepcidin analogue are expensive and are difficult to synthesize compared to the use of unlabeled standard. The intensities of Hepc-25 and ACTH in all samples had signal-to-noise of more than 70 : 1 with isotopic resolution of more than 13,000.
To determine that SPE breakthrough were not occurring, the eluant from the extraction step was re-introduced into another C-4 Omix tip and the extraction procedure was repeated. Subsequent analysis confirmed that hepcidin was absent following the second extraction.
Hepcidin retention on the C-4 sorbent bed during the ‘washing’ step (with 30% MeOH) was established. The eluant obtained during the washing step of the first extraction was analyzed using MALDI-TOF MS and no hepcidin peaks were observed. This demonstrated that the C-4 Omix tip was able to retain all of the hepcidin in the urine sample onto its sorbent bed and was only eluted during the elution step with 80% ACN/H2O/1% TFA (v/v/v). An extraction recovery of more than 98% and 70% was achieved when hepcidin standards were spiked in water at pH 7 (n = 3) and in blank urine (n = 2) respectively.
Hepcidin can rapidly oxidized in normal atmospheric condition due to the methionine residue that reacts with ambient ozone to give a molecular mass of 2805 Da.
The methionine residue in hepcidin can rapidly oxidized in ambient ozone to give a molecular mass of 2805 Da. Using the rapid µSPE extraction method described above together with the rapid analysis using the MALDI-TOF MS resulted in minimal oxidation of hepcidin.
Spot-to-spot reproducibility, intra and inter-day precision assay of urinary hepcidin
Apart from random fluctuations, the relationship between instrumental response and concentration may be interfered by matrix effects.40,41 The employment of standardized sample preparation steps and the addition of internal standards can minimize interference factors whereas matrix effects, which affect the sensitivity of the analysis, can be reduced by the addition of standard of the analyte to the sample (also known as standard addition method).Spot-to-spot reproducibility of spiked urine with different concentrations of hepcidin was performed. Hepcidin concentrations of 2.5 to 12.5 fmol/µL were spiked into extracted urine sample together with 12.5 fmol/µL of internal standard (ACTH). The percentage variation range and averages for 62.5, 25, 6.25, 2.5 fmol/µL of hepcidin spiked urine and unspiked urine are shown in Table 1. The spot-to-spot reproducibility percentage RSD was pooled from the all data from the intra- and inter-day analysis. These results showed that variations between spots with the same concentrations are below 6.5% with an overall average variation of less than or equal to 3.4% throughout the whole set. The set of urine samples with no spiking of hepcidin gave an average variation of only 3.1%.
The endogenous hepcidin concentration between sets ranged from 40.2 to 56.2 fmol/µL of urine. Intra-day (n = 5 sets) analysis over the range of 2.5 to 62.5 fmol/µL of spiked hepcidin into extracted urine and unspiked urine showed standard deviation range of 4.0 to 4.6 fmol/µL with a RSD of 9.0 to 9.5%.
Inter-day precision assay (n = 3 days), on the other hand, gave a standard deviation of 4.5 fmol/µL with a RSD of 9.4%. The correlation coefficient (R2) for all replicate sets was greater than 0.98, with an average (n = 18) of 0.99. The differences in peak area between the spiked hepcidin standard extract and the unspiked extract are displayed in Fig. 2.
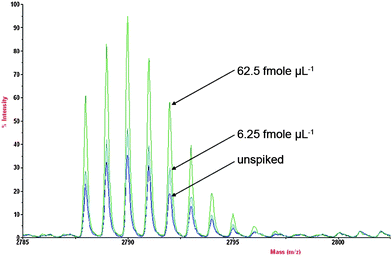 | ||
| Fig. 2 Endogenous hepcidin spiked with various concentration of hepcidin standards (62.5, 6.25 fmole µL−1 and unspiked) showing the differences in peak area. | ||
Creatinine analysis
A relative standard deviation of less than 0.5% was achieved between replicates and a R2 of more than 0.99 was obtained for creatinine analysis. With the diphenyl column, baseline separation of creatine and creatinine was achieved without the need for any sample pre-treatment with creatine and creatinine eluting at 3.0 min and 5.3 min respectively. The inter-day precision assay (n = 3 days) for creatinine was 1.69%.Relationship of urinary hepcidin with creatinine
Other groups have shown that urinary hepcidin level is usually normalized to urinary creatinine level.11,12,18,25 A RSD of 9.4% was achieved when hepcidin (nmol) was related to creatinine (mmol) giving a range of 2.2 to 2.7 nmol of hepcidin per mmol of creatinine with an average of 2.5 nmol of hepcidin/mmol creatinine. This data was consistent with the observations of other studies which reported between 0.4 to 2.2 nmol hepcidin/mmol creatinine.10,12,42 Ganz et al.18 postulated that 95% of hepcidin is retained in the kidney since hepcidin is not freely filtered through the glomerular membrane, and like other small peptides, hepcidin could also be reabsorbed and degraded in the proximal tubules which explained the low levels of hepcidin found in urine when compared to serum hepcidin levels.Limits of detection and stability tests
A signal-to-noise ratio of more than 20: 1 with isotopic resolution of more than 11,000 was obtained when 1 fmol of hepcidin standard together with 10 fmol of ACTH was spiked into the eluting buffer. This method showed that the monoisotopic resolution of hepcidin was possible even at extremely low levels.From the stability test (Fig. 3), Hepc-25 was stable for at least 7 hours at 30 °C. An exponential decrease in hepcidin concentration was observed after 7 hours of incubation (30 °C) where the bioactive hepcidin was degraded. Hepcidin did not appear to be oxidized after 7 hours of incubation as there was no observed increase in the oxidized hepcidin product at 2805 m/z. Hepcidin levels were relatively consistent following multiple freeze-thawing cycles (Fig. 4). Samples which underwent only 1 freeze-thaw cycle produced the highest hepcidin concentration. This result correlated with the findings by Kemna et al.24
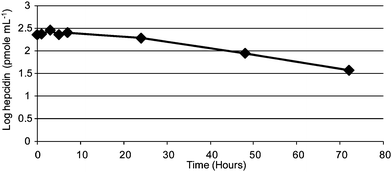 | ||
| Fig. 3 Stability of hepcidin-25 at 30 °C incubation for a total of 72 hours. | ||
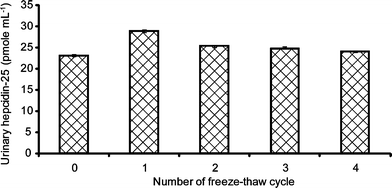 | ||
| Fig. 4 Influence of multiple freeze-thaw cycles on urinary hepcidin-25 reproducibility. | ||
When compared with other recent hepcidin studies (Table 2), the hepcidin level obtained in this study correlated well with the findings by Swinkels et al.,25 Nemeth et al.15 and Bansal et al.29 The normalization of hepcidin to creatinine levels will be an important reference tool for the study of other diseases such as hemochromatosis and sport-related disorder in relation to urinary hepcidin levels.
| Method | SELDIa | SELDI | Immuno-dot | LC/MSMSa | MALDI | MALDI |
|---|---|---|---|---|---|---|
| a Serum samples. | ||||||
| References | Tomosugi26 | Swinkels25 | Nemeth15 | Murphy30 | Bansal29 | This report |
| LOD (hepcidin/mL of sample) | 16 pmol/mL serum | NA | NA | 0.36 pmol/mL serum | NA | 1.25 pmol/mL urine |
| LOD (wrt creatinine (Cr)) | NA | 3–37 pmol/mmol Cr | 430 pmol/mmol Cr | NA | NA | 80.5 pmol/mmol Cr |
| Intra-day assay | 7.5–9.2% | 6.1–7.3% | 5–19% | N.D | 7–13% | 9.0–9.5% |
| Inter-day assay | 25.7–27.5% | NA | 12% | 11.0–15.3% | 10–20% | 9.4% |
The limit of detection (1.25 pmol/mL urine) obtained from this study showed comparable detection limits with serum hepcidin analysis performed by Murphy et al.30 (0.36 pmol/mL serum) and Tomosugi et al.26 (16 pmol/mL serum) using triple quadrupole MS and SELDI-TOF MS respectively. The limit of detection of hepcidin normalized to urinary creatinine obtained in our study was 80.5 pmol/mmol creatinine which was comparable with Swinkels et al.25 and Nemeth et al.15 with 3–37 pmol/mmol creatinine and 430 pmol/mmol creatinine respectively. The average urinary hepcidin levels reported by Bansal et al.29 was 10 nmol/mmol creatinine in healthy controls which is similar to our findings of an average of 2.4 nmol/mmol creatinine.
The precision assays for both intra- and inter-day assay obtained in this study showed RSD values of less than 10%, with an intra-day variation of only 0.5% compared with other studies showing RSD variation ranges of more than 1.2% shown in Table 2.15,25,26,29,30
In summary, a validated assay using standard addition of unlabeled hepcidin for the quantitation of endogenous hepcidin. This method is able to produce comparable or better intra- and inter-day reproducibility and detection limits for the quantification of hepcidin.
Conclusion
We reported the first standard addition method for the quantification of endogenous hepcidin using MALDI-TOF MS with a simple µSPE clean-up that requires only a small amount of sample (100 µL). This method was able to provide monoisotopic resolution and signal-to-noise ratio greater than 70 : 1 for hepcidin analysis with minimal oxidation of hepcidin-25. The spot-to-spot reproducibility of hepcidin standard additions (2.5–62.5 fmol/uL) were less than 3.5%. Intra- and inter-day precisions were both under 9.5% with an average of 2.4 mmol of urinary hepcidin per mmol of creatinine. Urinary hepcidin was able to withstand 2 freeze-thaw cycles and the extraction/analysis of hepcidin is recommended to be carried out within 7 hours at 30 °C after collection.Acknowledgements
M.C.L. Gay was supported with a Murdoch University International Student Scholarship. D. Trinder is the recipient of the Gastroenterological Society of Australia Senior Research Fellowship. J.K. Olynyk is the recipient of a National Health and Medical Research Council of Australia Practitioner Fellowship. The MALDI-TOF Mass Spectrometer was purchased with ARC LIEF Grant (LE0775763). The authors also thank Richard Lipscombe from Proteomics International for helpful discussions.References
- C. H. Park, E. V. Valore, A. J. Waring and T. Ganz, J. Biol. Chem., 2001, 276, 7806–7810 CrossRef CAS.
- A. Krause, S. Neitz, H.-J. Magert, A. Schulz, W.-G. Forssmann, P. Schulz-Knappe and K. Adermann, FEBS Lett., 2000, 480, 147–150 CrossRef CAS.
- E. V. Valore and T. Ganz, Blood Cells, Mol. Dis., 2008, 40, 132–138 CrossRef CAS.
- E. Nemeth and T. Ganz, Annu. Rev. Nutr., 2006, 26, 323–342 CrossRef CAS.
- I. De Domenico, D. M. Ward, E. Nemeth, M. B. Vaughn, G. Musci, T. Ganz and J. Kaplan, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8955–8960 CrossRef CAS.
- I. De Domenico, D. M. Ward and J. Kaplan, J. Clin. Invest., 2007, 117, 1755–1758 CrossRef CAS.
- C. Pigeon, G. Ilyin, B. Courselaud, P. Leroyer, B. Turlin, P. Brissot and O. Loreal, J. Biol. Chem., 2001, 276, 7811–7819 CrossRef CAS.
- G. Nicolas, C. Chauvet, L. Viatte, J. L. Danan, X. Bigard, I. Devaux, C. Beaumont, A. Kahn and S. Vaulont, J. Clin. Invest., 2002, 110, 1037–1044 CAS.
- E. Nemeth, S. Rivera, V. Gabayan, C. Keller, S. Taudorf, B. K. Pedersen and T. Ganz, J. Clin. Invest., 2004, 113, 1271–1276 CAS.
- E. Kemna, P. Pickkers, E. Nemeth, H. V. d. Hoeven and D. Swinkels, Blood, 2005, 106, 1864–1866 CrossRef CAS.
- E. Nemeth, E. V. Valore, M. Territo, G. Schiller, A. Lichtenstein and T. Ganz, Blood, 2003, 101, 2461–2463 CrossRef CAS.
- G. Papanikolaou, M. Tzilianos, J. I. Christakis, D. Bogdanos, K. Tsimirika, J. MacFarlane, Y. P. Goldberg, N. Sakellaropoulos, T. Ganz and E. Nemeth, Blood, 2005, 105, 4103–4105 CrossRef CAS.
- C. Peyssonnaux, A. S. Zinkernagel, R. A. Schuepbach, E. Rankin, S. Vaulont, V. H. Haase, V. Nizet and R. S. Johnson, J. Clin. Invest., 2007, 117, 1926–1932 CrossRef CAS.
- J. P. Pinto, S. Ribeiro, H. Pontes, S. Thowfeequ, D. Tosh, F. Carvalho and G. Porto, Blood, 2008, 111, 5727–5733 CrossRef CAS.
- E. Nemeth, A. Roetto, G. Garozzo, T. Ganz and C. Camaschella, Blood, 2005, 105, 1803–1806 CrossRef CAS.
- K. R. Bridle, D. M. Frazer, S. J. Wilkins, J. L. Dixon, D. M. Purdie, D. H. G. Crawford, V. N. Subramaniam, L. W. Powell, G. J. Anderson and G. A. Ramm, Lancet, 2003, 361, 669–673 CrossRef CAS.
- G. Papanikolaou, M. E. Samuels, E. H. Ludwig, M. L. E. MacDonald, P. L. Franchini, M.-P. Dube, L. Andres, J. MacFarlane, N. Sakellaropoulos, M. Politou, E. Nemeth, J. Thompson, J. K. Risler, C. Zaborowska, R. Babakaiff, C. C. Radomski, T. D. Pape, O. Davidas, J. Christakis, P. Brissot, G. Lockitch, T. Ganz, M. R. Hayden and Y. P. Goldberg, Nat. Genet., 2004, 36, 77–82 CrossRef CAS.
- T. Ganz, G. Olbina, D. Girelli, E. Nemeth and M. Westerman, Blood, 2008, 112, 4292–4297 CrossRef CAS.
- H. Kulaksiz, S. G. Gehrke, A. Janetzko, D. Rost, T. Bruckner, B. Kallinowski and W. Stremmel, Gut, 2004, 53, 735–743 CrossRef CAS.
- A. Amini and E. Nilsson, J. Pharm. Biomed. Anal., 2008, 46, 411–417 CrossRef CAS.
- V. Gelfanova, R. E. Higgs, R. A. Dean, D. M. Holtzman, M. R. Farlow, E. R. Siemers, A. Boodhoo, Y.-W. Qian, X. He, Z. Jin, D. L. Fisher, K. L. Cox and J. E. Hale, Briefings Funct. Genomics Proteomics, 2007, 6, 149–158 Search PubMed.
- T. J. Griffin, S. P. Gygi, B. Rist, R. Aebersold, A. Loboda, A. Jilkine, W. Ens and K. G. Standing, Anal. Chem., 2001, 73, 978–986 CrossRef CAS.
- S. S. Rubakhin and J. V. Sweedler, Anal. Chem., 2008, 80, 7128–7136 CrossRef CAS.
- E. H. J. M. Kemna, H. Tjalsma, V. N. Podust and D. W. Swinkels, Clin. Chem., 2007, 53, 620–628 CrossRef CAS.
- D. W. Swinkels, D. Girelli, C. Laarakkers, J. Kroot, N. Campostrini, E. H. J. M. Kemna and H. Tjalsma, PLoS One, 2008, 3, e2706 CrossRef.
- N. Tomosugi, H. Kawabata, R. Wakatabe, M. Higuchi, H. Yamaya, H. Umehara and I. Ishikawa, Blood, 2006, 108, 1381–1387 CrossRef CAS.
- S. Altamura, J. Kiss, C. Blattmann, W. Gilles and M. U. Muckenthaler, Biochimie, 2009 Search PubMed.
- D. G. Ward, K. Roberts, P. Stonelake, P. Goon, C. G. Zampronio, A. Martin, P. J. Johnson, T. Iqbal and C. Tselepis, Proteome Sci., 2008, 6, 28 CrossRef.
- S. S. Bansal, J. M. Halket, J. Fusova, A. Bomford, R. J. Simpson, N. Vasavda, S. L. Thien and R. C. Hider, Rapid Commun. Mass Spectrom., 2009, 23, 1531–1542 CrossRef CAS.
- A. T. Murphy, D. R. Witcher, P. Luan and V. J. Wroblewski, Blood, 2007, 110, 1048–1054 CrossRef CAS.
- U. Kobold, T. Dulffer, M. Dangl, A. Escherich, M. Kubbies, R. Roddiger and J. A. Wright, Clin. Chem., 2008, 54, 1584–1586 CrossRef CAS.
- H. Li, M. J. Rose, L. Tran, J. Zhang, L. P. Miranda, C. A. James and B. J. Sasu, J. Pharmacol. Toxicol. Methods, 2009, 59, 171–180 CrossRef CAS.
- S. S. Bansal, J. M. Halket, A. Bomford, R. J. Simpson, N. Vasavda, S. L. Thein and R. C. Hider, Anal. Biochem., 2008, 384, 245–253.
- N. Murao, M. Ishigai, H. Yasuno, Y. Shimonaka and Y. Aso, Rapid Commun. Mass Spectrom., 2007, 21, 4033–4038 CrossRef CAS.
- E. Kemna, H. Tjalsma, C. Laarakkers, E. Nemeth, H. Willems and D. Swinkels, Blood, 2005, 106, 3268–3270 CrossRef CAS.
- M. Zabet-Moghaddam, E. Heinzle, M. Lasaosa and A. Tholey, Anal. Bioanal. Chem., 2006, 384, 215–224 CAS.
- Y. L. Li and M. L. Gross, J. Am. Soc. Mass Spectrom., 2004, 15, 1833–1837 CrossRef CAS.
- I. V. Chernushevich, A. V. Loboda and B. A. Thomson, J. Mass Spectrom., 2001, 36, 849–865 CrossRef CAS.
- F. Hillenkamp and J. Peter-Katalinic, MALDI MS: A practical guide to instrumentation, methods and applications, Wiley-VCH, Weinheim, 2007 Search PubMed.
- A. I. Gusev, W. R. Wilkinson, A. Proctor and D. M. Hercules, Anal. Chem., 1995, 67, 1034–1041 CrossRef CAS.
- R. Knochenmuss, F. Dubois, M. J. Dale and R. Zenobi, Rapid Commun. Mass Spectrom., 1996, 10, 871–877 CrossRef CAS.
- L. Roecker, R. Meier-Buttermilch, L. Brechtel, E. Nemeth and T. Ganz, Eur. J. Appl. Physiol., 2005, 95, 569–571 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |