Observations on comprehensive two dimensional gas chromatography coupled with flame photometric detection for sulfur- and phosphorus-containing compounds
Sung-Tong
Chin
,
Ze-Ying
Wu
,
Paul Douglas
Morrison
and
Philip John
Marriott
*
Australian Centre of Research on Separation Science, School of Applied Sciences, RMIT University, G.P.O. Box 2476V, Melbourne, Vic 3001, Australia. E-mail: philip.marriott@rmit.edu.au; Fax: +61 3 96391321; Tel: +61 3 99252632
First published on 18th January 2010
Abstract
A comprehensive two-dimensional gas chromatography (GC × GC) system has been coupled to dual channel flame photometric detection (FPD) and evaluated for its performance in the analysis of sulfur- and phosphorus-containing compounds. The detector flow, including hydrogen, air and nitrogen gas was adjusted to achieve the maximum response and peak symmetry during GC × GC analysis in the sulfur and phosphorus mode respectively. Peak asymmetry for S-mode ranged from 0.30 to 0.66 and so exhibited considerable tailing for all flame gas settings. The peak asymmetry and peak width at half height (which ranged from 246 to 384 ms) were both relatively sensitive towards detector gas flow settings. By contrast in P-mode, peak asymmetry was almost unaffected by flame gas settings, and peak widths at half height were relatively constant, ranging from 165 to 185 ms. Results showed that increased detector acquisition rate up to 200 Hz distinctly reduced the signal-to-noise ratio, without affecting the modulated peak width. Occurrence of sulfur peak tailing behaviour during GC × GC analysis restricted the upper limit of sulfur response linearity range. Detection limits were estimated to be 617 pg/s in S-mode and 45 pg/s in P-mode for GC × GC operation, which were higher than the value estimated for 1D GC-FPD operation. The compatibility of GC × GC with FPD has been successfully demonstrated for the separation of sulfur-containing organophosphorus (OP) pesticides in S2 and HPO modes, as well as the organosulfur (OS) compounds in diesel and kerosene samples in S2 mode.
Introduction
The highly selective and sensitive performance of the flame photometric detector (FPD) is well-known for the analysis of organosulfur (OS) and organophosphorus (OP) compounds in various sectors namely petrochemical, environmental, biological, industrial hygiene and food products applications. As compared to the other more complex and/or expensive selective detectors for this purpose such as the pulsed flame photometric detector (PFPD), sulfur chemiluminescence detector (SCD), atomic emission detector (AED), and nitrogen chemiluminescence detector, the single-flame FPD does not require a high level of expertise for calibration, tuning, and maintenance to achieve adequate performance for trace level quantification in most analytical laboratories.1–3 The detection by flame photometry for S- and P-compounds is based on the selected chemiluminescence of the electronically excited S2* and HPO* molecules that yield strong specific optical emission bands with maxima at 394 nm and 526 nm respectively, when exposed to a hydrogen-rich flame.4,5 The selectivity of FPD response for both sulfur and phosphorus compared with hydrocarbons can be up to 1 × 106![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, with the detection limit reported to be as low as 3.6 pg/s for sulfur and 60 fg/s for phosphorus.6
:1, with the detection limit reported to be as low as 3.6 pg/s for sulfur and 60 fg/s for phosphorus.6
Despite various operational advantages, the sensitivity of the FPD is dependent on the operating conditions which require precise control of the gas flow rates and temperature.5 Detection in the single-flame FPD is also fraught with several difficulties such as cross interference of signal between sulfur and phosphorus channels (the selected channel exhibits some response from the presence of the other element in the detector), as well as tendency to response quenching for S- and P-containing compounds that co-elute with large hydrocarbon peaks during the chromatographic analysis. Such quenching effects may cause temporary or real flame-outs, or alter the mechanism of formation of the chemiluminescent species, and eventually impedes the accuracy of compound identification and quantification. Since the hydrocarbon response is not recorded, the presence of co-eluting compounds that quench response will not be apparent. In order to overcome the hydrocarbon quenching problem, various column arrangements and leaner hydrogen-air flame mixing conditions were tested to minimize the amount of hydrocarbon materials entering the FPD detector or reduce the effect of quenching.7 Most important is the necessity to ensure well resolved sulfur or phosphorus compounds from the major organic substances prior to FPD analysis. This might imply the use of a multidimensional gas chromatography (MDGC) system will be of advantage for improved resolution. However, reports of use of FPD with MDGC are relatively sparse.
The recent development of comprehensive two dimensional gas chromatography (GC × GC) has significantly improved the analytical throughput of ultra-complex samples with respect to resolved peaks, and has been applied both qualitatively and quantitatively.8,9 To adequately record modulated peak pulses with width at baselines as small as 100 ms, detector considerations that include aspects of ultra-fast signal acquisition rate and transduction, as well as geometry adaptation to suit faster mass flux are essential for reliable GC × GC analysis. The coupling of GC × GC with various detection systems has been tested in prior studies.10–12 Flame ionisation detection (FID) and time-of-flight mass spectrometry (TOFMS) are the universal detectors that have been most widely used in GC × GC analysis, but the demand for detectors with speciation capabilities for halogens, N-compounds and others, including low-level sulfur and phosphorus detection will likely increase in the future as responses to regulations for environmental and quality control applications are directed to GC × GC. Coupling the technique of GC × GC with SCD has been implemented for analysis of petroleum crudes and products, but undesired peak tailing was noticed leading to increased peak widths and decreased resolution.13,14 Most FPDs today are commonly connected to conventional single dimensional GC, so it is of interest to this work to test the implementation of GC × GC combined with a conventional FPD, since it is inevitable that this detection system should be evaluated for suitability for GC × GC operation.
The present work aimed to study the system performance of GC × GC connected to FPD for the analysis of sulfur and phosphorus compounds. The detector response behaviour under different FPD parameters was evaluated for selected standard compounds (i.e. tributyl phosphate and diphenyl sulfide) using simultaneous S-mode and P-mode FPD, with a dual FPD-FID arrangement employed to investigate relative peak parameters (primarily peak width properties) against the classical FID response. The practical application of the system to samples including diesel, kerosene and organophosphorus and organosulfur pesticides was examined.
Materials and methods
Sample and standard preparation
A stock solution of diphenyl sulfide (99% purity; BDH), tributyl phosphate (99% purity; Aldrich) at 1 g/L were prepared in hexane and stored below 4 °C prior to appropriate dilution for testing. “Aged” samples of kerosene and diesel were sourced from the RMIT Applied Chemistry stores as representative samples that contained elevated levels of sulfur compounds (recent samples have greatly reduced S-content). A range of organophosphorus pesticide compounds in a single mixture each at 1 mg/L concentration were gifts from SGE international (Ringwood, Australia) and the State Chemistry Laboratory (Victoria, Australia). The following compounds were used, with designated numerical identifications: 1; demeton-S, 2; ethoprop, 3; dichlorphos, 4; phorate, 5; demeton-O, 6; diazinon, 7; disulfoton, 8; methyl parathion, 9; ronnel, 10; chlorpyrifos, 11; fenthion, 12; trichloronate, 13; stirophos, 14; tribufos, 15; tokuthion, 16; fensulfothion, and 17; sulprofos.Instrumentation
An Agilent 7890A model gas chromatography (Agilent Technologies, Burwood, Australia) equipped with a Flame Ionization Detector and a dual channel Flame Photometric Detector (Agilent part number: G3430-63241) was used throughout this work, with 99.999% purity hydrogen as the carrier gas. A Rxi-5ms 30 m × 0.25 mm × 0.25 μm film thickness (df) column and a Stabilwax microbore 1 m × 0.1 mm × 0.10 μm df column from Restek Corporation (Buckinghamshire, UK) were installed as the primary and secondary dimension respectively. The latter column was passed through a Longitudinal Modulated Cryogenic System, LMCS (Chromatography Concepts; Doncaster, Australia) and connected to a Y-press fit which split the effluent equally through two 30 cm × 0.10 mm lengths of deactivated fused silica tubing into the base of the FID and FPD detectors. Thus parallel detection of FPD and FID was conducted, with simultaneous acquisition in HPO and S2 modes for the FPD detector.Note that since an Agilent FPD was used here, data, results or observations may not be directly transportable to other FPD systems, especially those that incorporate dual flames, counter-current flames, or pulsed flame operation.
Standard compound analysis with GC × GC-FPD/FID
Analysis by 1D GC was conducted with 1 μL aliquot GC injections at 250 °C under splitless mode for 0.5 min, with constant carrier gas flow at 1.5 mL/min. The oven temperature was programmed from 50 °C (hold 1 min), ramped to 150 °C at 30 °C/min, and finally ramped to 180 °C at 3 °C/min. The FID was maintained at 250 °C, with gas flow of 30 mL/min hydrogen, 300 mL/min air and 30 mL/min nitrogen. A similar system configuration was used for the evaluation of GC × GC-FPD except the modulation period (PM) was operated at 4 s and hold time of 0.4 s in the release position while the modulation temperature TM was set at 70 °C and 90 °C for TBP and DPS compounds respectively. During GC × GC analysis, the FPD was maintained at 250 °C, however since the FPD is to be optimised, various gas flow conditions and acquisition frequencies were used for evaluation of the detector performance.Sample analysis with GC × GC-FPD/FID
Analysis of diesel and kerosene specimens as well as organophosphorus pesticides (OP) were conducted using the above instrument with the same column set. For diesel sample, 1 μL aliquot without dilution was injected into the GC inlet at 300 °C and a split ratio of 50:1 with 1.5 mL/min constant H2 flow while the oven was programmed at 100 °C for 1 min, then increased to 230 °C at 5 °C/min with TM of 40 °C and modulation period (PM) of 7.5 s. Meanwhile, kerosene sample of 1 μL was injected with a split ratio 30:1 at 250 °C and 1.5 mL/min constant H2 flow. The oven temperature was held at 80 °C for 1 min, then ramped at a rate of 3 °C/min to 230 °C. A 5 s PM setting with TM of 0 °C was employed. The FPD condition was set at 250 °C with 50 mL/min H2, 60 mL/min air and 30 mL/min nitrogen gas flow and 100 Hz acquisition rate for optimum sulfur detection in both samples. Conversely, 1 μL of OP compounds (about 0.5 to 1.0 ng) was injected under splitless mode at 270 °C with 1.0 mL/min constant H2 flow. The oven temperature was ramped from 50 °C (1 min hold) to 130 °C at 12 °C/min, then increased to 240 °C at 5 °C/min and held for 6 min. A PM setting of 5 s and TM at 20 °C was used. For maximum phosphorus detection, the FPD was operated at 250 °C with 80 mL/min H2, 110/min air and 50 mL/min N2 gas flow. Data acquisition was at 100 Hz. The high data rate used here is consistent with usual FID and NPD acquisition rates with GC × GC, and ensures sufficient data density to record the quantitative measure of a very fast peak.
The GC × GC data including peak area, peak width at half height, and peak height were generated using ChemStation software (Agilent Technologies). The acquired data were further processed using an in-house program and visualized as contour plots using Origin software (OriginLab Corporation, Northampton, MA, USA). 2D data presentation for sample analysis has been adjusted by variation in the modulation commencement time, in order to present the majority of the peaks within the same presentation panel rather than have them exhibit the effect of wrap-around.15 This is merely a presentation convenience, and does not imply that this solves wrap-around. Thus it is possible to commence the modulator at, for example, ±0.01–±0.04 min to shift the data vertically by ±0.6–3.6 s. Since the data conversion process commences at time t = 0.0, this has the effect of changing the vertical position of all peaks. This will be discussed in the text as appropriate.
Results and discussion
Characterisation of modulated peak response
In the modulation process, sulfur and phosphorus solutes were successfully trapped and remobilized to generate several pulses, with FPD peak width at half height (wh) less than 350 ms, according to detector gas flow conditions. The two primary criteria of interest when describing the performance of modulated GC × GC peaks are the peak width of the pulsed peak, and the height of the peak. Both of these can inform the suitability of the detector for GC × GC operation. Peak width is of critical importance when the peak capacity of the second dimension determines the ability to separate neighbouring peaks. For instance, it has been reported that the NPD detector response leads to peak broadening for N-containing species, and the most favourable arrangement of the detector was to have a wide-bore collector and an extended jet. A regular jet leads to further broadened peaks, and reduced resolution and height.16 An Agilent micro-ECD detector (maximum 50 Hz data acquisition) had to be operated at gas flows well in excess of those recommended for 1D GC operation, with high flow needed in order to reduce peak width (i.e. reduces residence time in the ECD chamber) to improve the resolution. This also reduces the peak response height, presumably due to dilution effects.17Highly compressed effluent peaks require correct establishment of detector operating conditions for maximum performance, when adapting the detector to GC × GC analysis. The influence of different gas flows on the FPD response characteristics was evaluated by injecting solutions of 20 ng DPS and TBP in 1 μL hexane solvent into the GC column set under a 100 Hz data acquisition rate. Table 1 depicts that a higher FPD response for DPS detection was generally obtained with leaner fuel gas flow, but hydrogen flows < 50 mL/min resulted in a relatively unstable flame that extinguished frequently and is therefore impractical for chemiluminescence analysis. Sufficient amount of combustible fuel-oxidizer gas has to be supplied for stabilizing the flame propagation within the detector chamber while leaner fuel gas produced a lower temperature flame and this may permit more favourable formation of the stimulated molecular emission that allows the generation of the S2* species.18 The O/H ratio at about 0.25 produced response optima, which was less than the value of the previous study5 probably due to the varied manufacture design of the detector. Moreover, Quincoces and Gonzalez19 reported that increasing air flow rate reduced the S2 generation from sulfur atoms by competitive SO formation, causing decrease in S2 chemiluminescent response. Thus, maximum response for sulfur detection was conferred by a lower H2 flow, and air flow at 50 mL/min and 60 mL/min respectively. From Table 1, it also is apparent that high air flows cause both peak broadening, and worse peak symmetry, and so flame conditions that lead to broad, asymmetric peaks are associated with high air content. Thus at H2 = 50, 60, and 70 mL/min, broadest peaks arise for 70, 80, and 100 mL/min air flow, and generally smallest asymmetry values (As; measured at 10% peak height, tailing edge/leading edge) coincide with the same flow settings of air. A lower stoichiometric ratio hydrogen/air flame was reported to only be sustained in a specifically fabricated FPD geometry such as a micro counter-current cell or by flame pulsation where heat losses were drastically reduced.20,21 A micro counter-current FPD has been optimised by Kendler et al.22 for adaptation of ultrafast GC separation that can be operated with extremely low H2 flow of 5 mL/min. Peaks width as narrow as 13 ms were reported for fast temperature programming of a 0.5 m microbore column.
| Sulfur detection (n = 3) | Phosphorus detection (n = 3) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gas supply | Detector responsea | Symmetry (Asb) | w h (ms) | Gas supply | Detector response a | Symmetry (Asb) | w h (ms) | ||||
| H2 | Air | N2 | H2 | Air | N2 | ||||||
| a Detector response reflects the total peak area of all modulated peaks. b Peak symmetry As = peak leading edge/peak trailing edge. c Data reflects the peak width at half height of the most abundant modulated peak. d Note that the FID peak width at half height for DPS is ∼240 ms, and for TBP is ∼130 ms, indicating the relative retentions of the two compounds on the second column. Both have good symmetry in the FID (see Fig. 1). | |||||||||||
| 50 | 50 | 30 | 68013 ± 6864 | 0.66 ± 0.10 | 246 ± 18 | 70 | 80 | 30 | 10467 ± 542 | 0.81 ± 0.03 | 177 ± 3 |
| 50 | 60 | 30 | 83818 ± 5082 | 0.55 ± 0.04 | 253 ± 10 | 70 | 90 | 30 | 11583 ± 164 | 0.81 ± 0.01 | 175 ± 5 |
| 50 | 70 | 30 | 78095 ± 3017 | 0.45 ± 0.01 | 279 ± 15 | 70 | 100 | 30 | 14884 ± 53 | 0.79 ± 0.01 | 175 ± 6 |
| 50 | 50 | 50 | 60779 ± 3823 | 0.58 ± 0.01 | 261 ± 15 | 70 | 80 | 50 | 12355 ± 368 | 0.80 ± 0.02 | 175 ± 16 |
| 50 | 60 | 50 | 70266 ± 7439 | 0.53 ± 0.01 | 265 ± 4 | 70 | 90 | 50 | 13746 ± 597 | 0.81 ± 0.02 | 177 ± 7 |
| 50 | 70 | 50 | 72635 ± 2173 | 0.41 ± 0.03 | 295 ± 5 | 70 | 100 | 50 | 15528 ± 303 | 0.79 ± 0.02 | 179 ± 8 |
| 50 | 50 | 70 | 49338 ± 11697 | 0.62 ± 0.14 | 245 ± 21 | 70 | 80 | 70 | 12239 ± 1086 | 0.79 ± 0.04 | 176 ± 7 |
| 50 | 60 | 70 | 64619 ± 3021 | 0.44 ± 0.03 | 283 ± 7 | 70 | 90 | 70 | 14294 ± 55 | 0.80 ± 0.04 | 185 ± 2 |
| 50 | 70 | 70 | 62170 ± 1897 | 0.43 ± 0.08 | 351 ± 31 | 70 | 100 | 70 | 14604 ± 1486 | 0.78 ± 0.02 | 183 ± 10 |
| 60 | 60 | 30 | 80228 ± 1038 | 0.55 ± 0.03 | 256 ± 5 | 80 | 90 | 30 | 13482 ± 110 | 0.79 ± 0.02 | 176 ± 2 |
| 60 | 70 | 30 | 68137 ± 5177 | 0.54 ± 0.06 | 279 ± 29 | 80 | 100 | 30 | 15241 ± 174 | 0.77 ± 0.02 | 177 ± 6 |
| 60 | 80 | 30 | 68123 ± 6774 | 0.39 ± 0.01 | 302 ± 4 | 80 | 110 | 30 | 16273 ± 120 | 0.75 ± 0.02 | 173 ± 8 |
| 60 | 60 | 50 | 67023 ± 6596 | 0.55 ± 0.07 | 274 ± 19 | 80 | 90 | 50 | 14412 ± 507 | 0.77 ± 0.02 | 175 ± 1 |
| 60 | 70 | 50 | 66149 ± 3656 | 0.42 ± 0.01 | 286 ± 6 | 80 | 100 | 50 | 16324 ± 139 | 0.78 ± 0.01 | 175 ± 3 |
| 60 | 80 | 50 | 64079 ± 2873 | 0.39 ± 0.07 | 334 ± 30 | 80 | 110 | 50 | 17477 ± 161 | 0.75 ± 0.03 | 177 ± 5 |
| 60 | 60 | 70 | 58223 ± 3505 | 0.42 ± 0.01 | 278 ± 3 | 80 | 120 | 50 | 17359 ± 180 | 0.75 ± 0.07 | 183 ± 19 |
| 60 | 70 | 70 | 58774 ± 487 | 0.37 ± 0.01 | 317 ± 3 | 80 | 90 | 70 | 15249 ± 220 | 0.77 ± 0.02 | 172 ± 6 |
| 60 | 80 | 70 | 55989 ± 1291 | 0.33 ± 0.02 | 348 ± 3 | 80 | 100 | 70 | 16209 ± 66 | 0.77 ± 0.01 | 173 ± 8 |
| 80 | 110 | 70 | 16780 ± 321 | 0.74 ± 0.02 | 171 ± 3 | ||||||
| 70 | 70 | 30 | 66345 ± 11090 | 0.47 ± 0.02 | 275 ± 9 | 90 | 100 | 30 | 13508 ± 125 | 0.78 ± 0.01 | 165 ± 8 |
| 70 | 80 | 30 | 69305 ± 4394 | 0.41 ± 0.03 | 290 ± 8 | 90 | 110 | 30 | 14549 ± 117 | 0.76 ± 0.02 | 171 ± 4 |
| 70 | 90 | 30 | 64748 ± 2045 | 0.36 ± 0.02 | 321 ± 9 | 90 | 120 | 30 | 15241 ± 173 | 0.76 ± 0.02 | 172 ± 6 |
| 70 | 100 | 30 | 57831 ± 166 | 0.30 ± 0.01 | 356 ± 12 | 90 | 100 | 50 | 14439 ± 285 | 0.78 ± 0.02 | 169 ± 6 |
| 70 | 70 | 50 | 67163 ± 1872 | 0.42 ± 0.01 | 283 ± 6 | 90 | 110 | 50 | 15272 ± 244 | 0.76 ± 0.02 | 168 ± 6 |
| 70 | 80 | 50 | 63694 ± 2704 | 0.42 ± 0.07 | 323 ± 28 | 90 | 120 | 50 | 15921 ± 114 | 0.75 ± 0.01 | 167 ± 6 |
| 70 | 90 | 50 | 58834 ± 1273 | 0.32 ± 0.01 | 340 ± 6 | 90 | 100 | 70 | 14111 ± 1360 | 0.77 ± 0.01 | 173 ± 1 |
| 70 | 100 | 50 | 53319 ± 1472 | 0.30 ± 0.01 | 384 ± 7 | 90 | 110 | 70 | 14794 ± 553 | 0.77 ± 0.01 | 170 ± 1 |
| 90 | 120 | 70 | 15927 ± 279 | 0.75 ± 0.01 | 175 ± 3 | ||||||
For TBP detection in phosphorus mode, maximum response was achieved at H2 flow of 80 mL/min with an air flow of 110 mL/min (O/H ratio of 0.29) given that higher flame temperature is preferred to obtain maximum HPO* molecular emission.18 In fact, the fuel gas consumption was found to be marginally greater than the manufacturer's recommendation (75 mL/min H2 and 100 mL/min air for single dimension GC analysis); suggesting that higher H2 with adequate air flow favour rapid compound decomposition and HPO excitation processes with sufficient hydrogen and oxygen reactants in the flame plasma under fast elution during GC × GC analysis.
The comparison of parallel detection operation for FID and FPD under 1D chromatography conditions with respect to peak widths at half height indicates relatively similar data. The FPD peak width for S2 mode was about 2.47–2.23 s over the range 1 mg/L to 10 mg/L DPS while the respective data for FID peak width was about 2.41–2.48 s. Thus there is no significant difference in wh values. Low mass of S suffers greater effect of tailing whereas the narrower width for higher mass of S is attributed to the expected narrower wh value for S2 mode reported by Marriott,5 as a consequence of the square law response. Respective data for TBP with FPD HPO mode were: 2.32–2.29 s for 1–10 mg/L and 2.29–2.26 s for FID. In this case there is effectively no difference in peak widths for FID and FPD HPO mode. For modulated peak response (Table 1) peaks widths for S and HPO modes are now quite different, apparently due to the additional tailing for the sharp peaks of S.
Cardwell and Marriott5 observed that sulfur response of FPD decreased markedly as the N2 inert gas increased. In the present study, it is noted that N2 makeup flow as low as 30 mL/min was applied in the Agilent FPD (Table 1) in order to reduce the tailing tendency of DPS modulated peaks, while maintaining the detector sensitivity and stability. When exposed to the hydrogen-rich flame, S-containing compounds are decomposed and converted to a variety of combustion products by many bimolecular reactions, such as H2S, S, S2, SO and SO2, as well as the anticipation of various carbon-sulfur containing species under the presence of carbon radicals in the flame.23 Hence, the excited state S2* species could result from several multi-body collision reactions. Being a cooling medium, excessive N2 flow in the Agilent's FPD chamber would have also delayed these complex reactions; and consequently may reduce the sensitivity and promote peak tailing effects. Due to the tendency of compound adsorption on the exposed stainless steel shroud at the FPD's jet tip, Barinaga and Farwell24 found that addition of a small glass chimney over the tip significantly minimized the peak tailing by improving the mixture flow streaming. Shibamoto25 patented a modified FPD nozzle tip for a Shimadzu detector made of quartz to prevent the adhesion of sample component onto that region while Thurbide and Hayward26 observed less peak tailing when a larger custom-made FPD tubing was used.
On the other hand, the phosphorus pulsed peak symmetry remained good, at a mean value of 0.8 which was likely due to the relatively less complex (straight forward) HPO* species conversion process. Higher N2 flow at 50 mL/min was apparently needed in order to facilitate compound excitation in the plasma without overheating and degrading the species.
Fig. 1 shows the chromatogram of modulated FPD and FID peaks for DPS and TBP respectively. An average wh of 253 ms and 177 ms was observed for the most abundant FPD peaks of DPS and TBP respectively (Table 1). Notably, narrower and better peak shape for sulfur compounds could be achieved with FPD as opposed to SCD, which was reported to produce a peak that was 5-fold as wide at base than the equivalent FID peak.14 A calculation of As using the data in this cited paper gives a value of 0.25 for the Sievers' SCD, and 0.15 for the Antek SCD systems that were tested. In the present work with the FPD S-mode, the best As value was 0.66, and the least was 0.30. Overall, best response and asymmetry were found with the lower H2 flow of 50 mL/min. The peak widths of both FPD modes were however relatively broader than that generated with the FID which were 243 ms and 132 ms for DPS and TBP respectively throughout the test. The FID signal measurement is commonly based on the level of decomposed carbon that is ionized instantaneously by hydrogen and oxygen radicals within the flame plasma.23 In contrast to FID, longer residence time may be required in FPD measurement for a series of collision and conversion reactions prior to S2* and HPO* molecular emission. Furthermore, the FID gas flow velocities are considerably higher than that of the FPD, and so the speed of the FID response may lead to reduced residence time and faster elimination of the compound and ionisation products from the sensing region. On increasing the air flow in the FPD flame, more SO and SO2 molecules could have formed from DPS, and then subsequently converted to S2*, which may cause band broadening and tailing. Such mechanism has not been definitively identified.
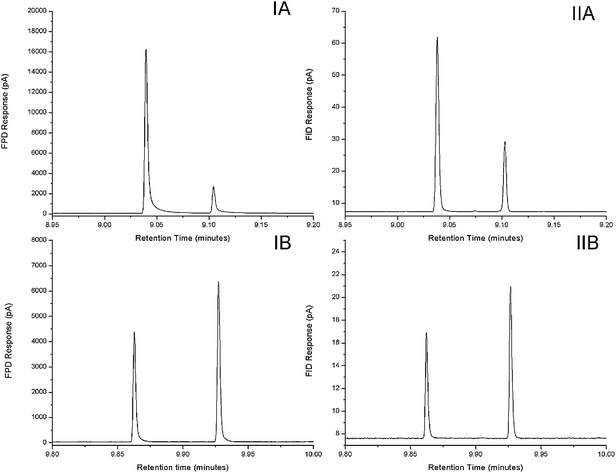 | ||
| Fig. 1 Modulated FPD (I) and FID (II) peak for (A) 7.5 ng injected masses of DPS, and (B) 5 ng injected masses of TBP. FPD-S mode was used for DPS, and FPD-P mode was used for TBP respectively at 100 Hz detector sampling rate. | ||
Alternatively the band broadening effect could also be caused by the excessive detector dead volume and adsorptive surfaces within the FPD chamber.24,26 Earlier works16,17 on the coupling of GC × GC with ECD and NPD also dictated that both detector cell volume and jet length play major roles in the modulated peak performance and minimizing the solute interaction within the chamber. Conditions had to be adopted that were outside the range of those recommended for each detector, in order to minimise peak asymmetry and maximise response. The duration of the S2* emission was found to be strongly correlated with the combustor length during flame pulsation in PFPD,27 whereby the emission duration was longest in a longer combustor tube. The specific characterization of PFPD in pulsed flame operation and the additional dimension of the time of generation of light emission provides considerable enhancement in detection sensitivity and selectivity over the single flame FPD used in this study by temporally separating the emission time of carbon species from that of sulfur and phosphorus.28 Nevertheless, suitability of PFPD to the fast data acquisition required in the GC × GC method has not been reported, and is likely to be unsuited to fast signal transduction (20 + Hz) as required in GC × GC. It is also of interest to note the region of the flame from which emission is stimulated. Different response processes and effects may arise if the stimulated response is from the central region of the flame, as opposed to the region about the periphery of the flame.25
Considering the response maxima and better peak symmetry, gas flows of 50 mL/min H2, 60 mL/min air and 30 mL/min N2 for sulfur detection mode and 80 mL/min H2, 110 mL/min air and 50 mL/min N2 for phosphorus detection mode were employed in the subsequent GC × GC study. Since the optimum settings are different for the two species, this means that simultaneous detection of both S-, P- and (S + P)-containing compounds in the one mixture will mean the detector flows should be set for either the best S response, or best P response, or set at a compromise condition.
Table 2 depicts the effect of data acquisition rate on the GC × GC-FPD detector response for both the sulfur and phosphorus mode, for DPS and TBP with average wh around 220 ms and 190 ms respectively. At 50 Hz sampling rate, over 20 data points are generated for the major effluent pulse which is sufficient for accurate peak description. Faster data acquisition rate, however, markedly increased the baseline noise; but concomitantly improved the response signal for both sulfur and phosphorus detection. The greater impact of noise on method sensitivity was reported to be determined by increasing sampling rate, increasing of peak width and decreasing of peak height.29 Although higher response can be obtained at faster sampling rate, the lower signal-to-noise ratio associated with 200 Hz would compromise trace compound analysis. Despite the maximum sampling rate of 500 Hz in the present FPD, the induced noise originating from the detector makes this frequency impractical. Apart from the optical filter, a finely tuned integrated noise filter network may be used in an attempt to minimize the high frequency noise. Multivariate techniques, such as Generalized Rank Annihilation Method (GRAM)30 and PARAllel FACtor analysis (PARAFAC),31 can be applied to extract the individual signals of the chemical components from that of interference and noise.
| Sampling rate | Sulfur detection (n = 3) a | Phosphorus detection (n = 3) b | ||||||
|---|---|---|---|---|---|---|---|---|
| Noise (pA) | Signal (pA) | S/N ratio | w h (ms) | Noise (pA) | Signal (pA) | S/N ratio | w h (ms) | |
| a Data reflects the mean value from 3 replicates tested using 750 pg DPS compound with FPD operated at 50 mL/min H2, 60 mL/min air, 30 mL/min N2. b Data reflects the mean value from 3 replicates tested using 100 pg TBP compound with FPD operated at 80 mL/min H2, 110 mL/min air, 50 mL/min N2. c Data indicated the peak width of the most abundant pulse peak. | ||||||||
| 50 Hz | 8 | 100 | 12.5 | 220 | 6 | 64 | 10.7 | 190 |
| 100 Hz | 11 | 150 | 13.6 | 220 | 7 | 72 | 10.3 | 190 |
| 200Hz | 16 | 120 | 7.5 | 220 | 12 | 76 | 6.3 | 190 |
The response calibration curve was evaluated by simultaneously and equally splitting the effluent from the column set to FID and FPD (Table 3). During GC × GC analysis, the FPD response was found to remain linear within the injected mass ranging from 0.05 to 50 ng for phosphorus detection, whilst S2* yielded a quadratic response over the mass amount ranging from 0.5 to 50 ng. The upper limit for sulfur response was restricted by half in contrast to GC-FPD analysis, which was attributed to unavoidable peak tailing at greater sulfur abundance. Furthermore, the slope value of the log (response) curve for both compounds shows a slight deviation, at around 1.3 and 1.7 for TBP and DPS respectively. Previous work 19 dictated that deviation of such value from linearity can affect the peak shape such as tailing, while the theoretical slope value can only be obtained with Gaussian peaks. Poole23 reported that such deviation is frequently observed in practice, which was contributed by the flame plasma fluctuation, compound-dependent decomposition, and competing flame reaction that lead to de-excitation.
| Sulfur mode a | Phosphorus mode b | |||||||
|---|---|---|---|---|---|---|---|---|
| Dynamic response range (ng) | Slope (log-log) | Noise (pA) | Minimum detection limit c (fg/s) | Response linearity (ng) | Slope (log-log) | Noise (pA) | Minimum detection limit c (fg/s) | |
| a Data shown as the detector response calibration range tested using DPS compound with FPD operated at 50 mL/min H2, 60 mL/min air, 30 mL/min N2. b Data shown as the detector response calibration range tested using TBP compound with FPD operated at 80 mL/min H2, 110 mL/min air, 50 mL/min N2. c MDL value with standard deviation was calculated using 5 calibration points ranging from 1 to 20 ng injection of DPS and TBP compound respectively. | ||||||||
| GC × GC-FPD | 0.5–50 | 1.77 | 11 | 617 ± 122 | 0.05–50 | 1.37 | 7 | 45 ± 17 |
| GC-FPD | 1–100 | 2.05 | 5 | 357 ± 82 | 0.1–50 | 1.18 | 4 | 23 ± 7 |
| GC × GC-FID | 0.1–100 | 0.98 | 0.1 | 82 ± 7 | 1–50 | 1.00 | 0.1 | 300 ± 105 |
The minimum detection limit for phosphorus mode in the present study is defined as MDLp = 2 × Np × mp/Ap, where Np is the noise, mp is the mass of phosphorus in the test substance and Ap is the total integrated area of the injected phosphorus compound.32 Calculation of MDL in sulfur mode is expressed as MDLs = [(2 × Ns × ms × ms)/(As × wh)]1/2, where Ns is the noise, ms is mass of sulfur in the test substance, As is the total integrated area of the injected sulfur compound, and wh is the peak width at half peak height.32 Although the actual detection sensitivity of both channels has been reduced due to effluent splitting, the result (Table 3) shows that the detection limit of GC × GC-FPD for HPO* species is insignificantly different to its value in 1D GC-FPD analysis; and still about 6 times lower than that with GC × GC-FID detection. Cryogenic modulation during GC × GC analysis did not improve the FPD detector sensitivity towards the S2* response, which exhibits a higher MDL value than 1D GC-FPD, probably due to high baseline noise caused by a fast detector sampling rate. Contrary to FID, the lower FPD sensitivity for sulfur detection arising from its non-linear response behaviour has been quoted previously by several workers.5,33 These studies also reported that a lower sulfur detection limit can be achieved by doping of a volatile trace sulfur compound through a permeation tube into the FPD detector, or in the H2 or air flow lines to linearize the response for low-level sulfur measurement. The detection limit in GC × GC analysis was previously improved by using GRAM to effectively correct the baseline offset.30
Application of GC × GC-FPD to OP and OS sample characterisation
This section will briefly survey the qualitative evaluation of the GC × GC-FPD method in P- and S-modes for typical sample analyses in petroleum and environmental (pesticide) studies. A quantitative study will be the subject of future work. OP pesticides are of continuing interest in many countries and monitoring programs for water and foods continues to be important. In the present work, separation of a 17 component OP pesticide compound mixture was used to illustrate the chromatogram and contour plots obtained with GC-FPD, GC × GC-FPD and GC × GC-FID respectively (Fig. 2). Under the P-mode configuration, the highly selective FPD detection readily distinguishes the distribution of OP compounds from other organic compounds that would otherwise interfere with the FID response, such as those derived from diluent solvents or 1D column bleed. Moreover, fensulfothion (peak 16), ronnel (peak 9), and dichlorphos (peak 3) were distinctly indicated by GC-FPD and GC × GC-FPD but not by GC × GC-FID. Fenthion (peak 11) which exhibited less favourable chromatographic behaviour in GC-FPD was clearly shown and well-separated from trichloronate (peak 12) in the contour plot acquired with both GC × GC-FPD and GC × GC-FID. Overall 2D peak widths at half height were observed to be < 0.3 s, apart from peak 11 which shows wrap-around in Fig. 2B, suggesting that OP compound detection by using the FPD with GC × GC is satisfactory.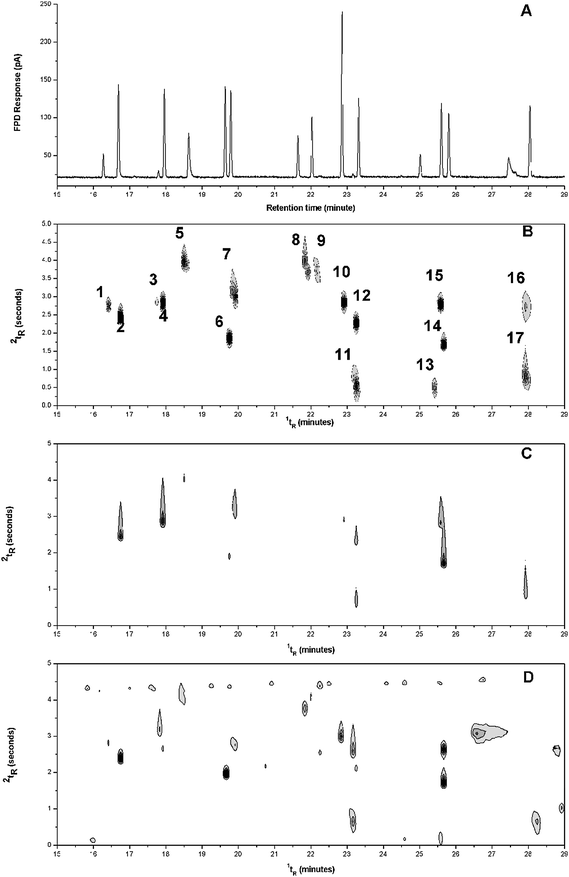 | ||
| Fig. 2 Gas chromatogram and contour plots of OP compounds at 1 mg/L obtained with (A) GC-FPD P-mode, (B) GC × GC-FPD P-mode, (C) GC × GC-FPD S-mode, and (D) GC × GC-FID. Refer to Experimental for compound designations. | ||
By contrast some peaks in FPD-S mode (Fig. 2C) show elongated tails, indicative of the tailing evident in Fig. 1IA. The organophosphate compounds contain various ratios of S, with one P atom. Compounds with no S atom (3, 13) have no response in S-mode. Compounds with one S atom (compounds 1, 5, 6, 8, 9, 10) generally have no, or poor response in the S-mode. Other OPs with 2 or 3 S atoms have varying response in FPD S-mode, and this testifies to the potential difficulty in assigning response factors to S-containing compounds in the single flame FPD. Fensulfothion (2 S atoms; peak 16) surprisingly appears to have no response. In contrast to the S-mode, the P-mode has a much more uniform response across the compound suite, with peaks in Fig. 2B having a much more consistent response as indicated by the contour plot. Peaks 3, 9, 13 and 16 appear to have reduced response, in both Fig. 2A and 2B, and so this suggests the standard mixture has degraded somewhat. This might account for the reduced fensulfothion response in the GC × GC FPD-S-mode. In Fig. 2B–D, the modulator start time was adjusted by +0.06 min, hence in Fig. 2D the band of bleed or trace non-polar compounds is located at about 4.3 s, instead of the expected ∼1.0 s. This brings most of the solutes into the same display panel, with only peaks 11, 13 and 17 located as wrap-around peaks, located at the lower part of the 2tR axis.
With respect to environmental and health related studies, sulfur speciation is an important quality assessment in the refinery process of petroleum products and in particular removal of the S-species from such products. Fig. 3 and Fig. 4 compare GC × GC-FPD and GC-FPD S-mode with GC × GC-FID for aged diesel and kerosene samples. Tentative identification of the compounds in the contour plot is referenced to results from previous study.14,34 In contrast to GC-FPD, additional information on the S-compound composition of diesel oil is revealed by GC × GC-FPD, for instance in its capacity to indicate related structural components due to the distinct patterns of related compounds in two-dimensional space.35 Thus, two different bands for the groups of benzothiophenes and dibenzothiophenes were located in a diagonal fashion as indicated in the contour plot, whilst within each cluster the component positions will vary according to alkyl chain length and extent of branching (Fig. 3B).
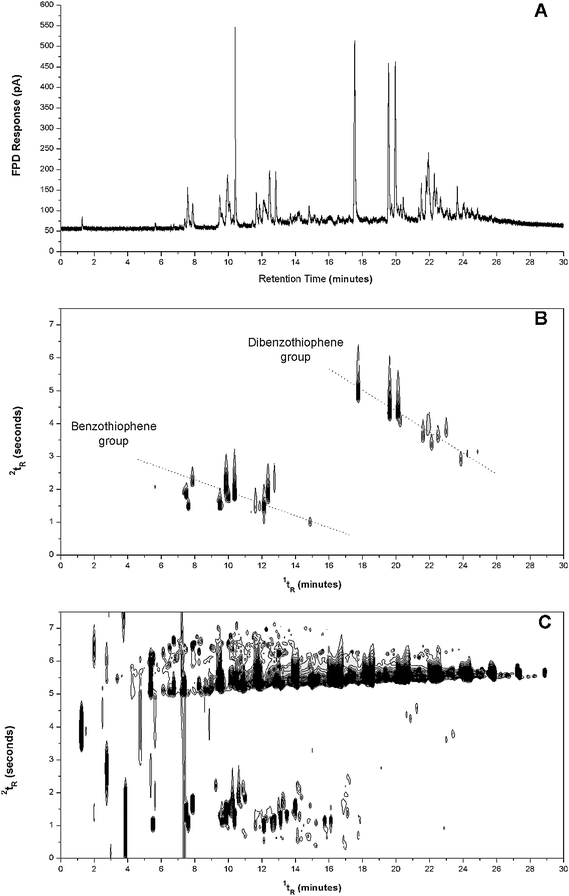 | ||
| Fig. 3 Gas chromatogram and contour plots of aged diesel obtained with (A) GC-FPD, (B) GC × GC-FPD, and (C) GC × GC-FID. FPD operated in S-mode. | ||
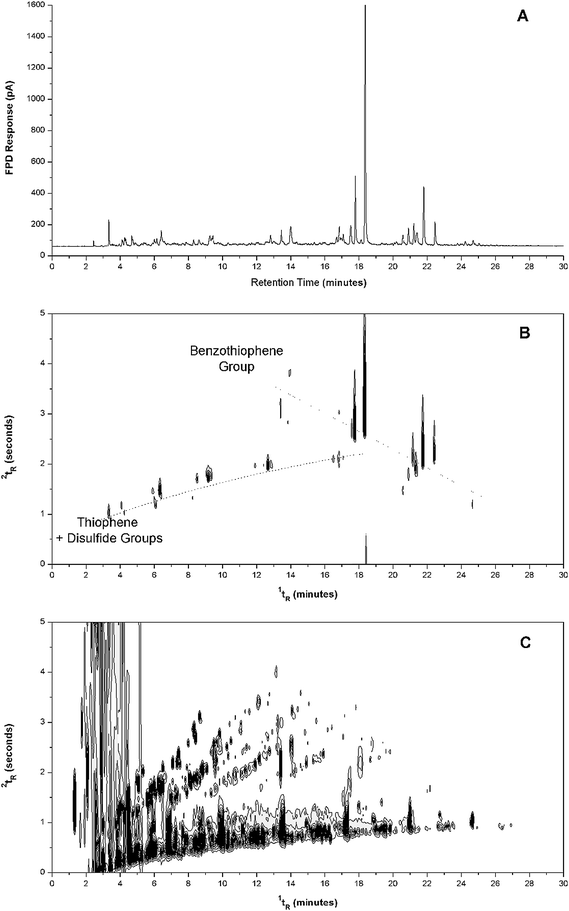 | ||
| Fig. 4 Gas chromatogram and contour plots of aged kerosene obtained with (A) GC-FPD, (B) GC × GC-FPD, and (C) GC × GC-FID. FPD operated in S-mode. | ||
Thiophene and benzothiophene clusters in kerosene were separated in 2D space (Fig. 4B) from other hydrocarbonaceous compounds detected by GC × GC-FID (Fig. 4C). Considerable peak tailing of large abundance sulfur peaks during modulation causes some wrap-around as seen in Fig. 4B. Note that even though the sulfur signal is very strong in FPD (both for GC and GC × GC) there is no corresponding peak shown at the presentation response level in the FID result (Fig. 4C). The abundant hydrocarbon peaks eluting at low 1tR in the GC × GC plot are not well trapped at the TM setting used, however as shown by the favourable sulfur compound peak shapes, these peaks are well defined as narrow focused spots. With relatively higher resolution efficiency (i.e. less peak broadening and tailing) compared to data shown in some reports of SCD coupled with GC × GC, hyphenation of GC × GC with FPD should offer a versatile and relatively lower cost alternative that will suit evaluation of the desulfurisation process, and other applications of S-compound analysis by using GC × GC. Note that Fig. 3 and 4 show the band of non-polar compounds located at ∼5.0 and 0.0 s respectively. This is done by adjustment of the modulator start time to be slightly later, and slightly earlier (by 0.04 and 0.02 min respectively) to bring the sulfur compounds into the same presentation space without apparent wrap-around. This can be seen in Fig. 3B and 4B respectively, where the S-compounds are displayed within the one panel. This is purely a convenience for presentation, and does not affect the modulation process.
The effect of potential quenching of response, especially for the S2 mode, is a cause for some comment. If S-compounds are now much better resolved, especially if they elute at a remote position in the 2D space away for hydrocarbon interferences, then an improved analysis should result. Since the present detector is a single flame detector, this may exacerbate the potential for quenching caused by overlapping hydrocarbons. Systematic study of such effects in GC × GC may be difficult to contrive, especially if peak overlap is difficult to achieve. However, perhaps also of interest will be to test dual-flame FPD systems, and the counter current FPD device. A pulsed flame FPD will be unlikely to achieve the data collection frequency desired for GC × GC operation.
Conclusion
Combination of GC × GC with FPD detection has been evaluated for suitability as a sensor for sulfur and phosphorus compounds, and applied to the characterization of OP and OS compounds in pesticides and petroleum products. The FPD parameters optimized included fuel, oxidant and inert gas flows, and sampling rate; these were adjusted for the fast solute detection required in GC × GC analysis. Relatively stable and reproducible response, combined with the simplicity of the detector with respect to the OP and OS compound detection was achieved. However, the symmetry of the narrow modulated sulfur peak is less than that of the compound detected by the FID detector, which was operated in parallel with the FPD detector. Whether this arises from the flame chemistry and structure, or is a consequence of the FPD geometry, supports the need for further study of FPD and research on possible improvements in detector design or flame structure to reduce the peak width for trace S-compound determination. By contrast, FPD in P-mode appears to be capable of fast and sensitive analysis of phosphorus compounds with acceptable peak symmetry and widths; in general the FPD-P mode has characteristics similar to that of FID in these respects. The FPD detection process for both S- and P-compounds is capable for very fast signal acquisition, and so is compatible with the fast peaks generated by the GC × GC experiment.Acknowledgements
The authors wish to acknowledge RMIT University for a postgraduate scholarship. Research funding support from the Australian Research Council under grant LP0776812 is acknowledged. Mr. Roy Hibbert (SGE International) and Mr. Gavin Rose (Victorian Department of Primary Industries) are thanked for the gift of pesticide standards.References
- R. L. Firor and B. D. Quimby, A Comparison of Sulfur Selective Detectors for Low Level Analysis in Gaseous Streams, Technical Report 5988-2426EN, Agilent Technologies, Wilmington, 2001 Search PubMed.
- K. K. Gaines, W. H. Chatham and S. O. Farwell, J. High Resolut. Chromatogr., 1990, 13, 489–493 CrossRef CAS.
- J.-P. Le Harle and B. Bellier, J. Chromatogr., A, 2005, 1087, 124–130 CrossRef CAS.
- P. J. Marriott and T. J. Cardwell, Chromatographia, 1981, 14, 279–284 CAS.
- T. J. Cardwell and P. J. Marriott, J. Chromatogr. Sci., 1982, 20, 83–90 CrossRef.
- Agilent 7890A Network Gas Chromatograph Data Sheet, Technical Note 5989-6317EN, Agilent Technologies, Wilmington, 2009 Search PubMed.
- J. Efer, T. Maurer and W. Engewald, Chromatographia, 1990, 29, 115–119 CrossRef CAS.
- O. Amador-Munoz and P. J. Marriott, J. Chromatogr., A, 2008, 1184, 323–340 CAS.
- J. Dalluge, J. Beens and U. A. T. Brinkman, J. Chromatogr., A, 2003, 1000, 69–108 CrossRef CAS.
- C. von Muhlen, W. Khummueng, C. A. Zini, E. B. Caramao and P. J. Marriott, J. Sep. Sci., 2006, 29, 1909–1921 CrossRef CAS.
- H. J. Tobias, G. L. Sacks, Y. Zhang and J. T. Brenna, Anal. Chem., 2008, 80, 8613–8621 CrossRef CAS.
- R. A. Shellie and P. J. Marriott, Analyst, 2003, 128, 879–883 RSC.
- J. Blomberg, T. Riemersma, M. van Zuijlen and H. Chaabani, J. Chromatogr., A, 2004, 1050, 77–84 CrossRef CAS.
- R. Ruiz-Guerrero, C. Vendeuvre, D. Thiebaut, F. Bertonini and D. Espinat, J. Chromatogr. Sci., 2006, 44, 566–573 CAS.
- R. C. Y. Ong and P. J. Marriott, J. Sep. Sci., 2002, 40, 276–291 CAS.
- C. von Muhlen, E. C. de Oliveira, P. D. Morrison, C. A. Zini, E. B. Caramao and P. J. Marriott, J. Sep. Sci., 2007, 30, 3223–3232 CrossRef CAS.
- E. M. Kristenson, P. Korytar, C. Danielsson, M. Kalliod, M. Brandt, J. Mäkelä, R. J. J. Vreuls, J. Beens and U. A. T. Brinkman, J. Chromatogr., A, 2003, 1019, 65–77 CrossRef CAS.
- J. Sevcik, Detectors in Gas Chromatography, Elsevier Scientific, Amsterdam, 1975 Search PubMed.
- C. E. Quincoces and M. G. Gonzalez, Chromatographia, 1985, 20, 371–375 CAS.
- K. B. Thurbide and C. D. Anderson, Analyst, 2003, 128, 616–622 RSC.
- G. Frishman and A. Amirav, Field Anal. Chem. Technol., 2000, 4, 170–194 CrossRef CAS.
- S. Kendler, S. M. Reidy, G. R. Lambertus and R. D. Sacks, Anal. Chem., 2006, 78, 6765–6773 CrossRef CAS.
- C. F. Poole, The Essence of Chromatography, Elsevier, Amsterdam, 2003 Search PubMed.
- C. J. Barinaga and S. O. Farwell, J. High Resolut. Chromatogr., 1986, 9, 474–476 CAS.
- United States Pat., US 2006/0213875 A1, 2006.
- K. B. Thurbide and T. C. Hayward, Anal. Chim. Acta, 2004, 519, 121–128 CrossRef CAS.
- S. Cheskis, E. Atar and A. Amirav, Anal. Chem., 1993, 65, 539–555 CrossRef CAS.
- A. Amirav and H. Jing, Anal. Chem., 1995, 67, 3305–3318 CrossRef CAS.
- W. A. Spencer and L. B. Rogers, Anal. Chem., 1980, 52, 950–959 CrossRef CAS.
- C. G. Fraga, B. J. Prazen and R. E. Synovec, J. High Resolut. Chromatogr., 2000, 23, 215–224 CrossRef CAS.
- J. M. Amigo, T. Skov and J. Coello, TrAC, Trends Anal. Chem., 2008, 27, 714–725 CrossRef CAS.
- P. Larson, D. Snyder, M. Abdel-Rahman, S. Engel, W. Wilson and T. Stark, Calculation of Performance Factors for Agilent 6890 Detectors Using Different Data Handling Devices, Technical note 5965-8901E, Agilent Technologies, Wilmington, USA, 1997 Search PubMed.
- G. W. Cobourn and R. E. Mulrooney, Atmos. Environ., 1988, 22, 1941–1947 CrossRef.
- F. C.-Y. Wang, W. K. Robbins, F. P. Di Sanzo and F. C. McElroy, J. Chromatogr. Sci., 2003, 41, 519–523 CAS.
- P. J. Marriott, T. Massil and H. Hugel, J. Sep. Sci., 2004, 27, 1273–1284 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |