Determination of atmospheric particulate-phase polycyclic aromatic hydrocarbons from low volume air samples†
Juana Mari
Delgado-Saborit
,
Noel
Aquilina
,
Stephen
Baker
,
Stuart
Harrad
,
Claire
Meddings
and
Roy M.
Harrison
*
Division of Environmental Health & Risk Management, School of Geography, Earth & Environmental Sciences, University of Birmingham, Edgbaston, Birmingham, UK B15 2TT. E-mail: r.m.harrison@bham.ac.uk; Fax: +44 (0)121 41 43709; Tel: +44 (0)121 41 43494
First published on 22nd January 2010
Abstract
This study has tested and optimized different filter media and pre-conditioning methods, extraction methodologies, cleaning techniques and solvents, concentration procedures and GC-MS parameters in order to establish the best methodology to sample and analyze particle-bound PAH collected in low volume samples (1.4 m3). The procedure developed combines the use of quartz fiber filters pre-conditioned at 400 °C for 48 h with a simple extraction procedure and optimized GC-MS parameters. The average method detection limits ranged from 4 to 15 pg m−3 for the 4–7 ring PAHs, precision (RSD) ranged from 0.3 to 9.7% and accuracy ranged from −6 to 25%. This method was validated with the extraction and analysis of the Standard Reference Material 1649a and was tested successfully on samples collected in outdoor microenvironments proving suitable for determination of particle-bound PAH concentrations without interferences in low volume samples.
1. Introduction
Polycyclic aromatic hydrocarbons (PAH) are a group of widespread environmental pollutants containing two or more fused benzene rings. PAHs are considered the most commonly distributed class of potent carcinogens present in the human environment, and many of them are listed as proven or possible carcinogens.1 Consequently, PAHs are widely studied with focus on their health-related impacts.2Atmospheric particle-bound PAHs are commonly sampled onto filter media by high-volume samplers collecting around 1000–2000 m3,3–5 while some authors have also used medium-volume samplers collecting between 7 and 30 m3.6,7 On the other hand, a few authors have used low volume samplers (<2 m3), with the analysis of these samples being performed with high performance liquid chromatography (HPLC)8,9 or in-line thermal desorption gas chromatography/mass spectrometry (GC-MS).10
Using low volume samples to analyse airborne PAH is vital for applications such as personal exposure assessment, where low sampling flowrates (e.g. 3 L min−1) are maintained for e.g. 24 h;11 for the study of diurnal variations, where snapshots of 1–2 h are required; or even for microenvironment characterization, where the lack of power supply requires the use of battery-operated equipment which can only maintain certain flowrates for short periods of time (e.g. 12 L min−1for 2 h).12
Sensitive, rapid and accurate methods have been developed to determine PAHs in atmospheric particles. As highly efficient separation tools, GC and HPLC have been used for analysing all kinds of samples containing complex components.13 While sensitive HPLC methods have been published for the determination of PAHs,14,15GC-MS is more commonly used due to greater separation efficiency of complex non-polar analytes.16 There are numerous standard procedures to determinate PAHs in ambient air using GC-MS, such as the EPA compendium method TO-13A,17 the Integrated Atmospheric Deposition Network to analyze PCBs, pesticides and PAHs in air and precipitation samples18 or the California Environmental Protection Agency method to determine PAHs in ambient air19 among others. However, all these methodologies collect PAHs in high volume samples and are not directly applicable to airborne particulate matter samples collected at low volume conditions. Low volume samples pose a challenge to analytical sensitivity. All atmospheric samples, i.e. high and low volume samples, are complex mixtures that contain a diverse range of substances. Hence, prior to analysis in a GC-MS, sample pretreatment is necessary to simplify the interpretation of chromatograms and mass spectra by preventing interfering compounds in the chromatograms. Typically, sample pretreatment for GC-MS involves three steps: extraction of the analytes, fractionation of the extracts by solid–liquid or liquid–liquid extraction, and since injection volumes of conventional GC-MS are small, evaporation of excess solvent to concentrate the analytes.20Extraction methods for PAHs from atmospheric samples include traditional Soxhlet,4,21 ultrasonic,22 microwave assisted,23 accelerated solvent,24 supercritical fluid25 and solid-phase microextraction.26 Whilst super fluid extraction and accelerated solvent extraction have high extraction efficiency, good selectivity and require low time for extraction, they require dedicated and more expensive equipment, which may sometimes preclude their application. On the other hand, traditional Soxhlet extraction is cheaper, but generates large amounts of solvents.13
Since low volume samples generally will contain small amounts of analyte, it is essential not only to reduce as much as possible the number of pre-treatment steps to reduce the level of blank contamination, but also to avoid the use of large solvent volumes which require subsequent concentration, hence increasing the risk of losing analytes by evaporation in the concentration steps.
This study has tested and optimized different GC-MS operational conditions, extraction procedures, cleaning techniques and solvents, different concentration methodologies, filter media and pre-conditioning methods in order to develop the best method able to sample and analyze particle-bound PAH collected in low volume air samples (1.4 m3) using low-cost extraction equipment and reducing as much as possible solvent use and sample handling. The optimized methodology for extraction and analysis was later validated with the Standard Reference Material 1649a. This method was used to measure snapshots of 2-h atmospheric samples in streets and other outdoor environments (i.e. parks).
2. Experimental
2.1. Atmospheric sampling
The particle-phase PAH measured and analysed were acenaphthylene (Ac), acenaphthene (Ace), fluorene (Fl), phenanthrene (Ph), anthracene (An), fluoranthene (Fluo), pyrene (Pyr), benz[a]anthracene (B[a]A), chrysene (Chry), benzo[b]fluoranthene (B[b]F), benzo[k]fluoranthene (B[k]F), benzo[a]pyrene (B[a]P), indeno[1,2,3-cd]pyrene (I[1,2,3-cd]P), benzo[ghi]perylene (B[ghi]P), dibenz[a,h]anthracene (D[a,h]A) and coronene (Cor).Particle-phase PAH were collected onto Membrane AQFA reinforced quartz fiber 47 mm filters (Millipore, Watford, UK), held in a polycarbonate filter holder, drawing air with a pump at a flowrate of 12 L min−1 for 2 h, collecting a final volume of 1.44 m3. Quartz fiber filters were pre-baked for 48 h at 400 °C. Samples were collected in different street microenvironments referred as trafficked roadsides, background streets, pedestrian streets and parks.
2.2. Reagents and standards
Dichloromethane (HPLC grade) was purchased from Fischer Scientific (Loughborough, UK) and nonane purum 99% was supplied by Sigma-Aldrich (Dorset, UK). Certified standard 16 EPA Priority PAH pollutant mixture CERTAN 100 μg/mL of each analyte in toluene was purchased from LGC Promochem (Teddington, UK). Coronene standard solution 100 μg mL−1 in toluene, acenaphthylene-d8 200 μg mL−1 in isooctane, pyrene-d10 500 μg mL−1 in acetone, chrysene-d12 2000 μg mL−1 in dichloromethane, benzo[a]pyrene-d12 200 μg mL−1 in isooctane, indeno[1,2,3-cd]pyrene-d12 200 μg mL−1 in isooctane, benzo[ghi]perylene-d12 200 μg mL−1 in toluene were supplied by Greyhound ChemService (Birkenhead, UK), benz[a]anthracene-d12 and phenanthrene-d10 1000 μg mL−1 in dichloromethane were purchased from UltraScientific (North Kingstown, RI, USA) whilst anthracene-d10 and p-terphenyl-d14 2000 μg mL−1 in dichloromethane were purchased from Greyhound ChemService and UltraScientific. Standard Reference Material SRM 1649a was supplied by Greyhound ChemService.The GC-MS system was calibrated with an eight calibration point curve. The concentrations level of the standards, which span the monitoring range of interest, were 0, 20, 50, 200, 500, 1000, 5000 and 10000 pg μL−1. All the standard solutions contained the internal standards at a concentration of 1000 pg μL−1. The recovery standard p-terphenyl-d14 was prepared at a concentration of 2000 pg μL−1.
2.3. Extraction, cleaning and concentration
PAH filters were spiked with a mixture of deuterated internal standards with concentration 1000 pg μL−1 dissolved in dichloromethane (DCM). Filters were placed in conical flasks with 15 mL of dichloromethane (HPLC grade) and shaken for 15 min at 1400 rpm using a reciprocating shaker. The extract was pre-concentrated to around 0.5 mL by blowing down with nitrogen and subsequently dried and cleaned by removing the remaining filter fibers with a chromatography column filled with 0.5 g of anhydrous sodium sulfate. The cleaned extract was then further concentrated by blowing down with nitrogen to 25 μL. The solvent was exchanged from DCM to nonane purum 99% with a final volume of 25 μL. Extracted samples were stored in GC vials in a freezer at −20 °C.Prior to analysis, every sample was spiked with 25 μL of the recovery determination standard (RDS), p-terphenyl-d14 to give a final extract volume of 50 μL. Samples were stirred to allow homogeneous mixing of the recovery standard with the sample using a vortexer.
2.4. Analysis of PAH samples
An Agilent Technologies 6890 Gas Chromatograph (GC) equipped with an Agilent HP-5MS, non-polar capillary column (30 m, 0.25 mm ID, 0.25 μm film thickness – 5% phenylpolysiloxane), in tandem with a 5973N Mass Spectrometer (MS) was used for the PAH analysis.1 μL of sample was injected using an Agilent 7683 auto-liquid sampler, in a splitless and non-pulsed injection mode at 300 °C. The initial temperature was held at 120 °C for 2 min and then ramped at 4 °C min−1 to a final temperature of 300 °C held for 10 min. The carrier gas was helium with a constant flowrate of 1 mL min−1. Solvent delay was set to 3.8 min.
The detector was set to quantify the analytes in single ion monitoring (SIM) mode covering specific masses ranging from 122 to 300 atomic mass units with a dwell time of 50 to 100 milliseconds per ion (Table 1). The selection of one target and one qualifier ion per compound proved sufficient for identification, whilst maximizing the time that the detector scanned each ion and hence improving the sensitivity of the SIM method. The mass spectrometer quad and source temperatures were 150 °C and 230 °C respectively. The analysis time per sample was 57 min.
| Compound | Target Ion (M)+ | Qualifier Ion (M)+ | Dwell Time (ms) | Retention Time (min) | Resolution Factor b | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Prog1 | Prog2 | Prog 3 | Prog4 | Prog1 | Prog2 | Prog 3 | Prog4 | ||||
| a n.m., not measured. b Resolution factors were calculated following the tangent method adopted by the United States Pharmacopeia (USP).53 c N/A, not applicable. | |||||||||||
| Acenaphthylene-d8 | 160.11 | 158.10 | 75 | 21.05 | 12.23 | 8.11 | 8.56 | 0.7 | 0.4 | 0.5 | 0.5 |
| Acenaphthylene | 152.06 | 151.06 | 75 | 21.09 | 12.27 | 8.15 | 8.61 | 7.9 | 7.4 | 8.0 | 8.8 |
| Acenaphthene | 153.07 | 154.08 | 100 | 21.64 | 12.79 | 8.84 | 9.40 | 14.6 | 20.8 | 21.4 | 21.9 |
| Fluorene | 166.08 | 165.07 | 100 | 23.17 | 14.24 | 10.80 | 11.70 | 22.7 | 32.7 | 33.2 | 33.5 |
| Phenanthrene-d10 | 188.14 | 184.11 | 75 | 26.01 | 17.02 | 14.72 | 16.39 | 0.6 | 0.7 | 0.8 | 0.8 |
| Phenanthrene | 178.08 | 176.06 | 75 | 26.08 | 17.09 | 14.81 | 16.50 | 0.7 | 0.8 | 1.0 | 1.2 |
| Anthracene-d10 | 188.14 | 184.11 | 75 | 26.16 | 17.17 | 14.93 | 16.65 | 0.4 | 0.6 | 0.6 | 0.7 |
| Anthracene | 178.08 | 176.06 | 75 | 26.21 | 17.23 | 15.00 | 16.74 | 22.8 | 40.9 | 43.2 | 47.1 |
| Fluoranthene | 202.08 | 200.06 | 75 | 29.74 | 20.70 | 20.19 | 23.10 | 2.9 | 8.3 | 7.3 | 6.9 |
| Pyrene-d10 | 212.14 | 208.11 | 75 | 30.32 | 21.32 | 21.06 | 24.16 | 0.4 | 0.7 | 0.6 | 0.6 |
| Pyrene | 202.08 | 200.06 | 75 | 30.40 | 21.38 | 21.14 | 24.25 | 18.1 | 39.7 | 10.8 | 11.0 |
| p-terphenyl-d14 | 244.39 | 122.20 | 100 | n.m.a | n.m. | 22.41 | 25.9 | n.m. | n.m. | 33.8 | 35.1 |
| Benzo(a)anthracene-d12 | 240.17 | 236.14 | 75 | 34.02 | 24.95 | 26.71 | 31.17 | 0.5 | 0.7 | 0.7 | 0.8 |
| Benzo(a)anthracene | 228.09 | 226.08 | 75 | 34.09 | 25.02 | 26.81 | 31.29 | 0.4 | 0.5 | 0.9 | 1.5 |
| Chrysene-d12 | 240.17 | 236.14 | 75 | 34.14 | 25.07 | 26.87 | 31.49 | 0.6 | 0.8 | 2.5 | 3.8 |
| Chrysene | 228.09 | 226.08 | 75 | 34.22 | 25.15 | 26.98 | 31.99 | 32.8 | 32.6 | 35.8 | 39.5 |
| Benzo(b)fluoranthene | 252.09 | 250.08 | 75 | 37.99 | 28.90 | 31.52 | 37.12 | 0.7 | 0.9 | 0.9 | 1.0 |
| Benzo(k)fluoranthene | 252.09 | 250.08 | 75 | 38.09 | 28.99 | 31.62 | 37.25 | 4.9 | 6.8 | 8.4 | 10.2 |
| Benzo(a)pyrene-d12 | 264.17 | 260.14 | 75 | 39.30 | 30.19 | 32.66 | 38.52 | 0.3 | 0.4 | 0.6 | 1.0 |
| Benzo(a)pyrene | 252.09 | 250.08 | 75 | 39.40 | 30.30 | 32.74 | 38.63 | 12.3 | 25.9 | 33.2 | 40.1 |
| Indeno(1,2,3-cd)pyrene-d12 | 288.17 | 284.14 | 50 | 45.53 | 36.38 | 36.79 | 43.65 | 0.3 | 0.8 | 0.7 | 0.7 |
| Indeno(1,2,3-cd)pyrene | 276.09 | 274.08 | 50 | 45.70 | 36.54 | 36.86 | 43.74 | 0.2 | 0.7 | 1.3 | 1.8 |
| Dibenz(a,h)anthracene | 278.11 | 276.09 | 50 | 45.86 | 36.72 | 37.03 | 43.97 | 1.9 | 5.3 | 5.2 | 5.2 |
| Benzo(ghi)perylene-d12 | 288.17 | 284.14 | 50 | 47.29 | 38.10 | 37.58 | 44.62 | 0.3 | 0.7 | 0.7 | 0.7 |
| Benzo(ghi)perylene | 276.09 | 274.08 | 50 | 47.47 | 38.28 | 37.66 | 44.72 | 14.1 | 28.0 | 31.9 | 36.1 |
| Coronene | 300.09 | 298.08 | 100 | 62.29 | 53.00 | 43.58 | 51.04 | N/Ac | N/A | N/A | N/A |
Each chromatogram was checked using MSDS Chemstation software. The samples were analyzed and quantified using a six-point calibration graph of the concentration ratio of analyte to internal standard against the corresponding peak area ratios using linear regression.
3. Results and discussion
3.1. Optimization of GC-MS conditions
Several GC columns, ramp rates, injection and initial temperatures as described in detail in Table 2 have been tested in order to establish the best conditions for which the internal and natural standards peaks were separated and identified.| Compound | Program 1 | Program 2 | Program 3 | Program 4 |
|---|---|---|---|---|
| Injection Temperature (°C) | 250 | 250 | 300 | 300 |
| Injection Mode | Splitless | Splitless | Splitless | Splitless |
| Initial GC Temperature (°C) | 40 | 120 | 120 | 120 |
| Initial GC Time (min) | 1 | 2 | 2 | 2 |
| Rate (°C min−1) | 8 | 8 | 5 | 4 |
| Final GC Temperature (°C) | 300 | 300 | 300 | 300 |
| Final GC Time (min) | 35 | 32 | 10 | 10 |
| Run Time (min) | 69 | 57 | 48 | 57 |
| GC Column | Varian CP-7950 | Varian CP-7950 | HP-5MS | HP-5MS |
| GC Column Dimensions | 60 m × 0.2 mm × 0.2 μm | 60 m × 0.2 mm × 0.2 μm | 30 m × 0.25 mm × 0.25 μm | 30 m × 0.25 mm × 0.25 μm |
| Flow (mL min−1) | 1 | 1 | 1 | 1 |
| Detector Temperature (°C) | 280 | 280 | 280 | 280 |
The starting conditions (Program 1) were those described by Lim et al. (1999).51 In brief, the program consists of an initial temperature of 40 °C, a ramping rate of 8 °C min−1 up to 300 °C using a 60 m Varian CP7950 DB5 (60 m, 0.2 mm id, 0.2 μm df) column. The injector mode was splitless non-pulsed, the injection and detector temperatures were both 300 °C, the carrier gas was helium at 1 mL/min and the GC-MS was set up in splitless mode.
The separation and resolution between peaks for some of the standards was poor (e.g.pyrene-d10 and pyrene, Fig. 1a), the peaks had low response and some of the peaks appeared with a shoulder (e.g.fluoranthene, Fig. 2a) and in some cases there was overlapping between consecutive peaks (e.g.indeno[1,2,3-cd]pyrene and dibenz[a,h]anthracene, Fig. 3a). A second program using the same column was tested which included raising the initial temperature from 40 °C to 120 °C. This second program had a run time shorter than the first, but the problem with close peaks, overlapping and shouldering of some peaks still persisted. The possibility that the shoulder represented a distinct peak was rejected as all the standard solutions were prepared with certified standards and therefore the compounds present in the mixture and the approximate retention times were known.
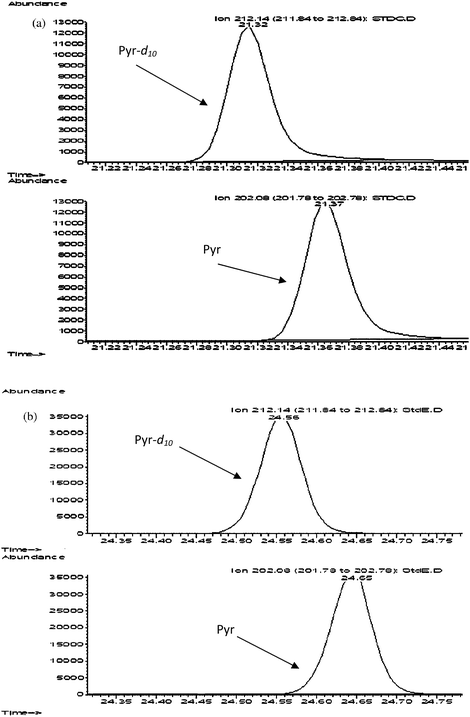 | ||
| Fig. 1 Retention time difference of two consecutive peaks e.g.pyrene-d10 (1st and 3rdchromatogram) and pyrene (2nd and 4thchromatogram) with (a) GC-MS Program 1 and (b) GC-MS Program 4. (a) Retention time difference between peaks: 0.05 min, resolution factor: 0.4. (b) Retention time difference between peaks: 0.09 min, resolution factor: 0.6. | ||
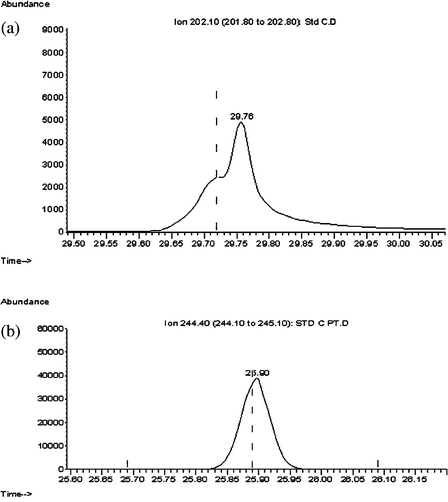 | ||
| Fig. 2 Peak shape and intensity e.g.fluoranthene (a) GC-MS Program 1 and (b) GC-MS Program 4. | ||
![Peak separation/overlap of two consecutive peaks e.g.Indeno[1,2,3-cd]pyrene (1st peak) and dibenz[a,h]anthracene (2nd peak) with (a) old GC-MS method and (b) new GC-MS method. (a) Retention time difference between peaks: 0.16 min, resolution factor: 0.2 and the peaks overlap. (b) Retention time difference between peaks: 0.23 min, resolution factor: 1.8 and the peaks do not overlap.](/image/article/2010/AY/b9ay00157c/b9ay00157c-f3.gif) | ||
| Fig. 3 Peak separation/overlap of two consecutive peaks e.g.Indeno[1,2,3-cd]pyrene (1st peak) and dibenz[a,h]anthracene (2nd peak) with (a) old GC-MS method and (b) new GC-MS method. (a) Retention time difference between peaks: 0.16 min, resolution factor: 0.2 and the peaks overlap. (b) Retention time difference between peaks: 0.23 min, resolution factor: 1.8 and the peaks do not overlap. | ||
A new set of GC-MS programs were then tested after changing the GC column to DP-5MS (30 m, 0.25 mm id, 0.25 μm df) and lowering the ramping rate to 5 °C min−1 (Program 3) and 4 °C min−1 (Program 4). The rest of the program parameters were maintained (i.e. injector mode, injector temperature, carrier flowrate and MS mode). In the new GC-MS program a considerable improvement was observed in peak separation with better resolution between peaks increasing the difference in the retention times of standard peaks by up to a factor of two (Fig. 1b), the resolution factor improved (Table 1), the peaks appeared well defined, the peak intensity was around ten-fold higher (Fig. 2b) and the problem of overlapping of peaks was solved (Fig. 3b).
Further optimization of the GC-MS program was performed checking the injection conditions by comparing the results of the splitless non-pulsed mode (Fig. 4a) with the splitless pulsed mode (Fig. 4b). Better chromatography results were obtained in the splitless non-pulsed mode program, with higher intensity of peaks and a better separation compared with the splitless pulsed mode program.
![Peak separation of two consecutive peaks e.g.benz[a]anthracene (first peak) and chrysene (second peak) with (a) pulsed injection and (b) non-pulsed injection.](/image/article/2010/AY/b9ay00157c/b9ay00157c-f4.gif) | ||
| Fig. 4 Peak separation of two consecutive peaks e.g.benz[a]anthracene (first peak) and chrysene (second peak) with (a) pulsed injection and (b) non-pulsed injection. | ||
The conditions set in Program 4 with splitless non-pulsed injection were selected as the most appropriate considering the better separation and resolution between peaks with respect to Program 3 (See Table 1).
The selected GC-MS program was used to develop and validate the method of extraction as well as to perform a blank contamination study in different filter media to see the best condition to sample and extract PAH from ambient air.
3.2. Optimization of extraction method
To test the recovery of the extraction method, blank filters were spiked with 50 μL of internal and natural standards at a concentration of 1000 pg μL−1 and were subsequently extracted using different extraction methods. Extracting filters spiked with standards was preferred over extracting certified materials for two reasons. The first was to homogenize the matrix as samples are collected onto filters and certified materials are normally powder. Secondly, the certified material 1649a was more expensive than the PAH standards solution. Therefore, employment of the standards for method development and the use of the certified materials for method validation as shown in Section 3.5 was preferred, considering the large number of tests performed to optimize the extraction method.Originally, samples were extracted with accelerated solvent extraction (Dionex ASE-200), pre-concentrated with the TurboVap (Zymark),27,28 cleaned with 10 mL of DCM through 0.5 g of Florisil inactivated, and were further concentrated to 25 μL by blowing a gentle stream of N2, to be finally solvent exchanged to nonane, following established procedures within our analytical group.29 However, the recovery efficiency of this methodology was around 70%. Therefore, different combinations of purificationsolvents (hexane, hexane/toluene (9.5/5.4 v/v), dichloromethane, hexane/DCM (6/4 v/v) at different volumes (10 mL to 150 mL) were tested in order to increase the recovery factor, giving results ranging from 25% to 76%. However, the more volatile standards (e.g.acenaphthylene-d8) had recoveries ranging from 3 to 72%. (Detailed information on extraction conditions tested and recovery factors can be found in Supporting Information.)†Accelerated solvent extraction has normally high efficiency recoveries.30 Therefore the poor performance of this proposed sequence of procedures to extract the PAHs (i.e.ASE followed by clean-up with Florisil) might be due to the large quantities of solvent used in the combination of extraction and clean-up steps that need to be evaporated, which implies a risk of losing analytes by evaporation as otherwise suggested by the low recoveries of the more volatile PAHs (see Supporting Information).†
To improve the recovery, especially for the more volatile standards, a simpler extraction method was tested. The proposed method consisted of the extraction of the PAH from the filters by shaking them in conical flasks with DCM, concentration of the sample by blowing N2 gently, cleaning the extract of fibres with a column of sodium sulfate anhydrous and further concentration of the extracts with an N2 stream before exchanging the solvent to nonane. (See specific details in the Experimental section.)
With this method, the recovery efficiency of both the internal and natural standards was much improved, with average recovery values of 106 ± 4%. This method was adopted as the extraction procedure not only because it was the one which had the highest recovery factors but also because this method involved less sample handling, less solvent use and therefore shorter extraction times, less risk of losing analytes by evaporation and less risk of high blank levels due to simplicity of the pre-treatment steps.
3.3. Optimization of filter media
As reported by many authors, glass fibre filters and quartz fibre filters have been used commonly to collect particle-bound PAHs.13 Considering that the methodology developed is aimed at analysing low volume samples with reduced amounts of analyte, reducing the blank introduced by the filter media is paramount. Some researchers have solvent-extracted31,32 or baked33–35 the filter media as a measure to lower the filter blank contamination. In this study different filter media (i.e. glass and quartz fibre filters) and different pre-conditioning methods have been tested in order to assess the best combination to reduce blank levels in the filter media.Glass (GFF) and quartz (QFF) fibre filter media were tested as received (RAW-treatment), after baking for 48 h in a Carbolite oven at 400 °C (48H-treatment) and for a set of glass fibre filters, thermal conditioning followed by extraction with DCM by accelerated solvent extraction (Dionex ASE-200) (48H-ASE-treatment). Results of each pre-conditioning treatment performed on 5 replicate filters spiked with 50 μL of internal standard 1000 pg μL−1 solution and subsequently extracted as described in the Experimental Section are presented in Table 3.
| Compound | GFF-RAW | GFF-48H | GFF-48H-ASE | QFF-RAW | QFF-48H | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mean | STD | Mean | STD | Mean | STD | Mean | STD | Mean | STD | |
| a GFF = Glass Fibre Filter, QFF = Quartz Fibre Filter, RAW = No pre-treatment, 48H = Baked 48 h at 400 °C, ASE = Cleaned with DCM in the ASE, STD = Standard deviation. | ||||||||||
| Acenaphthylene | 93.3 | 57.7 | 32.0 | 27.6 | 74.1 | 74.7 | 2.4 | 0.8 | 1.7 | 0.1 |
| Acenaphthene | 523.3 | 207.7 | 455.2 | 522.3 | 396.0 | 283.9 | 7.0 | 1.9 | 9.1 | 0.9 |
| Fluorene | 598.5 | 221.1 | 600.4 | 564.1 | 1154.1 | 1231.7 | 11.0 | 5.3 | 9.7 | 1.7 |
| Phenanthrene | 19.3 | 2.8 | 20.2 | 7.6 | 22.8 | 5.5 | 10.0 | 2.1 | 11.8 | 1.4 |
| Anthracene | 0.3 | 0.1 | 0.5 | 0.1 | 0.8 | 0.6 | 0.9 | 0.3 | 2.8 | 5.3 |
| Fluoranthene | 84.1 | 28.8 | 1.1 | 0.2 | 1.4 | 0.6 | 1.5 | 0.7 | 1.1 | 0.3 |
| Pyrene | 15.8 | 32.3 | 0.9 | 0.1 | 1.3 | 0.7 | 1.5 | 0.4 | 0.9 | 0.1 |
| Benzo(a)anthracene | 0.3 | 0.1 | 0.3 | 0.1 | 0.7 | 0.6 | 0.3 | 0.2 | 0.3 | 0.1 |
| Chrysene | 0.8 | 0.1 | 0.7 | 0.1 | 1.0 | 0.7 | 0.4 | 0.2 | 0.6 | 0.2 |
| Benzo(b)fluoranthene | 0.5 | 0.0 | 0.4 | 0.1 | 0.8 | 0.7 | 0.3 | 0.2 | 0.5 | 0.1 |
| Benzo(k)fluoranthene | 0.6 | 0.1 | 0.5 | 0.0 | 1.0 | 1.1 | 0.4 | 0.2 | 0.5 | 0.1 |
| Benzo(a)pyrene | 0.6 | 0.1 | 0.7 | 0.1 | 1.4 | 1.2 | 0.8 | 0.2 | 0.6 | 0.1 |
| Indeno(1,2,3-cd)pyrene | 1.0 | 0.1 | 1.4 | 0.3 | 1.9 | 1.6 | 1.3 | 0.3 | 1.1 | 0.2 |
| Dibenz(ah)anthracene | 0.5 | 0.2 | 0.6 | 0.1 | 1.2 | 1.3 | 0.4 | 0.1 | 0.5 | 0.1 |
| Benzo(ghi)perylene | 1.0 | 0.1 | 1.0 | 0.1 | 2.2 | 2.2 | 1.1 | 0.1 | 1.0 | 0.1 |
| Coronene | 0.2 | 0.1 | 0.4 | 0.4 | 0.5 | 0.2 | 0.1 | 0.1 | 0.2 | 0.1 |
Glass fibre filters showed higher blank levels for the more volatile PAH compounds (i.e.acenaphthylene to phenanthrene) compared with the quartz fibre filters (t-test for comparison of means, p < 0.05), although for the rest of the compounds (i.e. high molecular weight PAH), the levels were comparable. In addition, those results from the thermal followed by the extraction treatment (48H-ASE-treatment) showed the highest blank levels throughout all the PAH compounds (p < 0.05). Hence, quartz fibre filters were preferred as a filter medium. As regards the comparison between different pre-conditioning techniques for quartz fibre filters, baking the filters at 400 °C for 48 h lowered the blank levels for the more volatile PAHs (p < 0.05), whilst the levels for the remaining PAH compounds were very similar between the RAW- and the 48H-treatments. In view of the data, quartz fibre filters thermally pre-conditioned at 400 °C for 48 h were selected as the appropriate filter media to sample particle-bound PAH in low volume samples.
3.4. Recovery levels, limits of detection and precision of the method proposed
The average recovery of the spiked standards across the whole range of tested concentrations (Table 4) was 89 ± 8% for the internal standards and up to 104 ± 15% for the natural standards. Looking into the different standards, the lowest recoveries were recorded for the more volatile compounds i.e.acenaphthylene-d8, phenanthrene-d10, anthracene-d10 and acenaphthylene with recovery percentages ranging 70–80%. Despite this, the efficiency of the proposed method is considered suitable and is in the same range as those reported elsewhere2,35–37 for GC-MS analysis of airborne PAH collected in high- and medium-volume samples.
| STD A 20 pg μL−1 | STD B 50 pg μL−1 | STD C 200 pg μL−1 | STD D 500 pg μL−1 | STD E 1000 pg μL−1 | Average | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | STD | Mean | STD | Mean | STD | Mean | STD | Mean | STD | Mean | STD | |
| Internal Standard Recoveries | ||||||||||||
| Acenaphthylene-d8 | 70 | 6 | 72 | 6 | 72 | 12 | 77 | 5 | 79 | 4 | 74 | 7 |
| Phenanthrene-d10 | 77 | 4 | 80 | 4 | 79 | 11 | 85 | 5 | 85 | 4 | 81 | 6 |
| Anthracene-d10 | 78 | 6 | 81 | 4 | 81 | 11 | 86 | 5 | 86 | 4 | 82 | 7 |
| Pyrene-d10 | 86 | 5 | 88 | 4 | 87 | 11 | 93 | 4 | 92 | 4 | 89 | 7 |
| Benzo(a)anthracene-d12 | 91 | 9 | 93 | 5 | 91 | 12 | 100 | 5 | 98 | 4 | 95 | 8 |
| Chrysene-d12 | 92 | 7 | 88 | 4 | 91 | 13 | 100 | 11 | 100 | 11 | 94 | 10 |
| Benzo(a)pyrene-d12 | 91 | 12 | 95 | 5 | 94 | 13 | 102 | 4 | 100 | 3 | 96 | 9 |
| Indeno(1,2,3-cd)pyrene-d12 | 88 | 15 | 93 | 5 | 93 | 14 | 104 | 5 | 102 | 2 | 96 | 11 |
| Benzo(ghi)perylene-d12 | 87 | 13 | 93 | 4 | 93 | 13 | 103 | 4 | 100 | 2 | 95 | 10 |
| Natural Standard Recoveries | ||||||||||||
| Acenaphthylene | 78 | 17 | 85 | 6 | 79 | 12 | 82 | 5 | 83 | 4 | 81 | 10 |
| Acenaphthene | 106 | 12 | 93 | 9 | 86 | 13 | 87 | 5 | 89 | 4 | 88 | 21 |
| Fluorene | 110 | 7 | 102 | 4 | 91 | 14 | 92 | 6 | 94 | 4 | 94 | 22 |
| Phenanthrene | 149 | 7 | 118 | 7 | 94 | 12 | 94 | 5 | 93 | 5 | 110 | 23 |
| Anthracene | 87 | 3 | 89 | 4 | 88 | 12 | 93 | 5 | 94 | 4 | 90 | 7 |
| Fluoranthene | 104 | 6 | 104 | 5 | 102 | 13 | 109 | 5 | 112 | 4 | 106 | 8 |
| Pyrene | 105 | 5 | 99 | 5 | 95 | 12 | 100 | 5 | 100 | 4 | 100 | 7 |
| Benzo(a)anthracene | 107 | 8 | 101 | 5 | 99 | 13 | 107 | 5 | 109 | 4 | 105 | 8 |
| Chrysene | 92 | 4 | 95 | 5 | 101 | 13 | 110 | 6 | 111 | 4 | 102 | 11 |
| Benzo(b)fluoranthene | 101 | 11 | 104 | 5 | 111 | 16 | 132 | 7 | 137 | 4 | 117 | 17 |
| Benzo(k)fluoranthene | 93 | 12 | 96 | 4 | 108 | 15 | 121 | 6 | 124 | 4 | 108 | 15 |
| Benzo(a)pyrene | 100 | 10 | 100 | 5 | 102 | 14 | 112 | 5 | 114 | 4 | 106 | 10 |
| Indeno(1,2,3-cd)pyrene | 108 | 9 | 106 | 5 | 103 | 15 | 116 | 5 | 119 | 2 | 110 | 10 |
| Dibenz(ah)anthracene | 100 | 13 | 99 | 7 | 112 | 18 | 138 | 7 | 149 | 2 | 116 | 33 |
| Benzo(ghi)perylene | 106 | 8 | 103 | 3 | 104 | 15 | 116 | 5 | 117 | 3 | 109 | 9 |
| Coronene | 108 | 14 | 109 | 6 | 120 | 19 | 152 | 8 | 163 | 2 | 126 | 36 |
| Average Internal & Natural Standard Recoveries | 97 | 9 | 95 | 5 | 95 | 13 | 104 | 6 | 106 | 4 | 99 | 13 |
The instrument detection limit (IDL), defined as the amount of pollutant that gives a signal to noise ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, was determined by calculating the signal to noise ratio for the pollutant in the lowest calibration standard (in our case 20 pg μL−1). The sample detection limit (SDL) was calculated considering the final extract volume (50 μL), the sample size (1.44 m3) and the percentage recovery of internal standard (Table 4) used to quantify the target pollutant in a particular sample.38 The method detection limit (MDL) was calculated as three times the standard deviation of the blank determination (i.e.quartz fiber filter pre-baked at 400 °C for 48 h).
:1, was determined by calculating the signal to noise ratio for the pollutant in the lowest calibration standard (in our case 20 pg μL−1). The sample detection limit (SDL) was calculated considering the final extract volume (50 μL), the sample size (1.44 m3) and the percentage recovery of internal standard (Table 4) used to quantify the target pollutant in a particular sample.38 The method detection limit (MDL) was calculated as three times the standard deviation of the blank determination (i.e.quartz fiber filter pre-baked at 400 °C for 48 h).
Table 5 shows the instrument, sample and method detection limits obtained with the proposed methodology. The instrument detection limits are better than those reported by some workers16,39 and similar to those reported by some others.2,40 As regards the sample and method detection limits, these are considerably lower than the respective limits of detection reported previously for PAHs in ambient air37,41 which may be attributed to the combination of better instrument sensitivity, lower filter blank levels due to the preconditioning of the filter and lower contamination levels throughout the extraction, cleaning and concentration of the sample prior to GC-MS analysis.
| Compound | Instrument Limit of Detection | Sample Detection Limit | Method Detection Limit | Accuracy (%) a | Precision (%) a | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Average (pg μl−1) | STD (pg μl−1) | Average (pg m−3) | STD (pg m−3) | Average (pg m−3) | STD (pg m−3) | Average (%) | STD (%) | Average (%) | STD (%) | |
| a Accuracy and precision calculated from filters spiked with standard solution. See Table 6 for values of accuracy and precision calculated from the extraction of SRM 1649a. | ||||||||||
| Acenaphthylene | 1.0 | 1.0 | 28.1 | 29.0 | 90.2 | 347.6 | 5.4 | 5.5 | 0.7 | 0.4 |
| Acenaphthene | 5.0 | 5.9 | 152.8 | 176.3 | 488.4 | 1821.0 | 13.7 | 7.2 | 2.2 | 1.1 |
| Fluorene | 5.0 | 6.7 | 163.2 | 198.9 | 609.8 | 2082.0 | 21.8 | 11.7 | 3.9 | 2.7 |
| Phenanthrene | 6.7 | 7.2 | 255.9 | 131.5 | 79.0 | 65.1 | 16.1 | 17.5 | 2.6 | 3.3 |
| Anthracene | 1.4 | 2.0 | 43.8 | 38.2 | 281.3 | 379.2 | 2.5 | 1.5 | 0.8 | 0.5 |
| Fluoranthene | 0.6 | 0.7 | 22.1 | 11.3 | 15.1 | 16.8 | 3.6 | 1.5 | 0.7 | 0.8 |
| Pyrene | 0.5 | 0.5 | 17.4 | 7.5 | 9.9 | 1.6 | 2.5 | 2.0 | 1.2 | 0.7 |
| Benzo(a)anthracene | 0.2 | 0.2 | 7.3 | 2.1 | 6.8 | 7.1 | 2.7 | 1.2 | 0.4 | 0.1 |
| Chrysene | 0.3 | 0.4 | 10.6 | 5.4 | 13.2 | 11.9 | 7.4 | 2.8 | 7.4 | 1.1 |
| Benzo(b)fluoranthene | 1.0 | 0.8 | 40.6 | 12.1 | 5.7 | 5.2 | 9.4 | 11.2 | 7.3 | 1.2 |
| Benzo(k)fluoranthene | 0.3 | 0.4 | 11.3 | 5.0 | 3.9 | 3.2 | 25.1 | 8.4 | 9.7 | 2.5 |
| Benzo(a)pyrene | 0.3 | 0.4 | 11.0 | 5.7 | 8.0 | 4.9 | −3.3 | 2.5 | 0.3 | 0.1 |
| Indeno(1,2,3-cd)pyrene | 0.6 | 0.8 | 22.9 | 10.7 | 13.9 | 11.2 | −6.0 | 1.8 | 1.1 | 1.7 |
| Dibenz(a,h)anthracene | 0.3 | 0.4 | 12.1 | 4.9 | 4.1 | 0.4 | 4.5 | 13.5 | 1.4 | 0.7 |
| Benzo(ghi)perylene | 0.7 | 0.4 | 26.5 | 2.5 | 8.0 | 3.7 | 0.2 | 1.8 | 0.7 | 0.8 |
| Coronene | 0.1 | 0.2 | 4.4 | 1.7 | 4.4 | 1.4 | 4.7 | 14.3 | 2.1 | 0.7 |
The limits of detection of the proposed method were also compared with those reported by Gil-Molto et al. (2009),10 who collected low-volume samples but analyzed them with in-port thermal desorption instead. The instrument limits of detection for the particle-bound PAHs (i.e.B[a]A to Ind) obtained with the present procedure were two-fold lower than the methodology developed by Gil-Molto et al. (2009).10 As regards the method and sample detection limits, these were 2 to 3-fold lower in the proposed methodology compared with the quantification detection limits of these authors (Gil-Molto et al. 2009).10 Other authors that have used in-port thermal desorption with high-volume samples reported considerably higher limits of detection than those obtained with the proposed methodology.42,43
Precision was calculated as the relative standard deviation (i.e. 100 × σn−1/average) of concentrations obtained from the 5 replicate analyses of the same sample at each concentration ranges (i.e. 20 pg μL−1 to 1000 pg μL−1) and the overall average values are presented in Table 5. The average precision of the method is 3.2 ± 1.9% (arithmetic mean (AM) ± standard deviation (STD)) which is comparable with values reported in the literature.16
The accuracy of the method was calculated as the difference between the true value of the quantity being measured (concentration spiked) and the result of the measurement (concentration analysed), normalized by the true value times 100. The present method has an average accuracy of 13.4 ± 17.3% (AM ± STD), ranging between −6% and 25%. The highest values (i.e. poorer accuracy) corresponded with compounds that did not have their own internal standard, as in the cases of Ace, Fl and B[k]F with average accuracy values ranging 14–25%. Phenanthrene had also high values of accuracy (16.1 ± 17.5%), which could be a consequence of the proximity of the peak of anthracene, which makes correct separation of both peaks difficult on some occasions and hence increases the accuracy value. However, experimental concentration means of the spiked filters extracted were compared with the nominal standard concentration values using the ANOVA test (SPSS 15.0). None of the compounds had statistically significant differences between the nominal and analyzed values (p > 0.1). Similarly, the precision of extracted spiked filters was compared with the precision of the analysis of the standard solutions and no statistically significant differences were found (p > 0.10).
The values of accuracy and precision of this method accomplish the quality objectives for air toxics stated by the EPA in the “Quality Assurance Handbook for Air Pollution Systems” which should be a precision of ±25% and an accuracy of ±20%.44 Only benzo[k]fluoranthene shows values of accuracy above the limit of the requirement.
3.5. Standard reference material analysis
Five samples (1 mg) of the Standard Reference Material NIST SRM 1649a – (Urban Dust) were analyzed in order to validate the accuracy and the precision of the method. Experimental concentrations were compared with the certified concentration using a one-sample t-test (SPSS 15.0). Most of the compounds did not present statistically significant differences between the certified and analyzed values (p > 0.10), with the exception of An, B[k]F and D[ah]A (p < 0.05) and Chry (p < 0.01), which had higher concentrations in the experimental dataset. Those compounds were the ones showing higher values of accuracy and precision, ranging from 32–50% and 22–38% respectively, in contrast with the low accuracy and precision values of the majority of the compounds (i.e. 0.2–8.7% and 4–18% respectively). In summary, the mean experimental concentrations obtained in this study when the SRM was treated, extracted and analyzed in the same way as the proposed extraction method were generally consistent with the certified values of concentrations (Table 6). Similar results were reported by Crimmins and Baker16 for the high molecular weight PAH, whilst better reproducibility and accuracy are reported in this study for the low molecular weight compounds (i.e.phenanthrene to fluoranthene).| Compound | Certified Concentration (mg kg−1) | Certified Variability (mg kg−1) | Mean Experimental Concentration (mg kg−1) | Standard Deviation (mg kg−1) | Precision (%) (RSD) | Accuracy a (%) |
|---|---|---|---|---|---|---|
| a Expressed as (mean measured concentrations minus certified value)/Certified Value × 100. b Certified and experimental concentration are significantly different at p < 0.05 level. c Certified and experimental concentration are significantly different at p < 0.01 level. | ||||||
| Phenanthrene | 4.140 | 0.370 | 4.50 | 0.73 | 16.2 | 8.7 |
| Anthracene | 0.432 | 0.082 | 0.58b | 0.16 | 27.5 | 34.3 |
| Fluoranthene | 6.450 | 0.180 | 6.75 | 1.24 | 18.3 | 4.7 |
| Pyrene | 5.290 | 0.250 | 5.49 | 0.90 | 16.4 | 3.8 |
| Benzo(a)anthracene | 2.208 | 0.073 | 2.15 | 0.40 | 18.4 | −2.6 |
| Chrysene | 3.049 | 0.060 | 4.03c | 0.90 | 22.3 | 32.2 |
| Benzo(b)fluoranthene | 6.450 | 0.640 | 6.74 | 1.21 | 17.9 | 4.5 |
| Benzo(k)fluoranthene | 1.913 | 0.031 | 2.85b | 1.08 | 38.0 | 49.0 |
| Benzo(a)pyrene | 2.509 | 0.087 | 2.47 | 0.10 | 3.9 | −1.6 |
| Indeno(1,2,3-cd)pyrene | 3.180 | 0.720 | 3.17 | 0.28 | 8.9 | −0.3 |
| Dibenz(ah)anthracene | 0.288 | 0.023 | 0.40b | 0.14 | 35.3 | 38.9 |
| Benzo(ghi)perylene | 4.010 | 0.910 | 4.00 | 0.34 | 8.6 | −0.2 |
3.6. Concentrations of particulate-phase PAH in ambient air
After validating the methodology with the NIST SRM 1649a standard, low volume samples collected in outdoor air in different street microenvironments were extracted and analyzed for particulate-bound PAH using the method described in this study.The average concentration of benzo[a]pyrene measured as a marker of the carcinogenic activity of the PAH mixture, was 0.09 ng m−3 in parks, 0.18 ng m−3 in pedestrian streets, 0.16 ng m−3 in background streets and 0.26 ng m−3 on trafficked roadsides. The minimum benzo[a]pyrene value reported in outdoor air was 0.05 ng m−3 which is 7.5 times the Method Detection Limit, showing the suitability of the proposed method to sample and analyze particle-bound PAHs in low volume and low concentration samples.
The air quality in Birmingham seems to have improved as evidenced by the concentrations of particulate-phase B[a]P, 0.48 ng/m3 measured in Birmingham urban air (1996)45 and 0.26 ng/m3 in Birmingham trafficked roadside (2008). This trend is consistent with previously reported studies which indicated a decrease of PAH levels in ambient air in Germany46 and USA.47 The B[a]P values in this study are comparable though generally lower than typical values obtained elsewhere in Europe in the last decade, which range from 0.7–2.97 ng/m3.48–50
In street microenvironments, traffic is a major source of PAH, and therefore, it would be expected that the magnitude of PAH concentrations would correlate with traffic volumes. A t-test was performed comparing the values measured at trafficked roadside locations with PAH concentrations measured away from traffic. The results of PAH concentrations for compounds An-Chry, B[k]F, B[a]P and B[ghi]P (Fig. 5) show that PAH concentrations measured in trafficked streets were generally higher (p < 0.005) compared with other street types, with parks being the outdoor environment where the lowest PAH concentrations were generally recorded. This is consistent with traffic loads as reported by previous studies.51,52
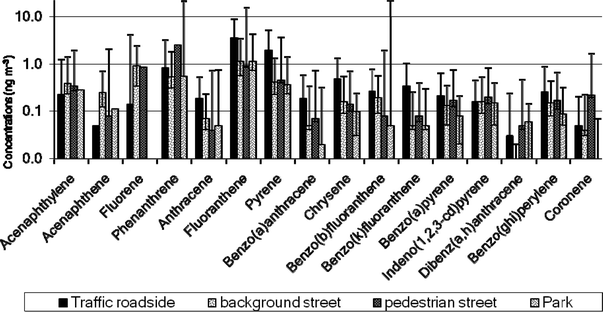 | ||
Fig. 5
PAH in different street microenvironments. (NTrafficked roadside = 8 ( ), Nbackground street = 10 ( ), Nbackground street = 10 ( ), Npedestrian street = 4 ( ), Npedestrian street = 4 ( ), Npark = 4 ( ), Npark = 4 ( )). The bars represent the 5–95%ile range of the distribution of values. )). The bars represent the 5–95%ile range of the distribution of values. | ||
4. Conclusion
This study has tested different filter media and pre-conditioning methods, extraction methodologies, cleaning techniques and cleaning solvents, concentration procedures and GC-MS conditions in order to establish the best methodology to sample and analyze particle-phase PAH collected in low volume samples.The methodology developed, which combines optimized GC-MS parameters, a simple extraction procedure with the use of quartz fiber filters pre-conditioned at 400 °C for 48 h has not only been successfully characterized (i.e. detection limits, precision and accuracy) but also has been validated with the analysis of a Standard Reference Material.
Moreover, the analysis of specially collected atmospheric samples has shown the suitability of the proposed method to determine PAH concentrations without interference in real samples. The proposed methodology has therefore been demonstrated to be suitable to sample and analyze particle-bound PAHs collected in low volume samples (1.44 m3).
| Ac | Acenaphthylene |
| Ace | Acenaphthene |
| AM | Arithmetic mean |
| An | Anthracene |
| B[a]A | Benz[a]anthracene |
| B[a]P | Benzo[a]pyrene |
| B[b])F | Benzo[b]fluoranthene |
| B[ghi]P | Benzo[ghi]perylene |
| B[k]F | Benzo[k]fluoranthene |
| Chry | Chrysene |
| Cor | Coronene |
| D[a,h]A | Dibenz[a,h]anthracene |
| DCM | Dichloromethane |
| Fl | Fluorene |
| Fluo | Fluoranthene |
| GC–MS | Gas chromatography mass spectra |
| HPLC | High performance liquid chromatography |
| I[1,2,3-cd]P | Indeno[1,2,3-cd]pyrene |
| IDL | Instrument detection limit |
| MDL | Method detection limit |
| PAH | Polycyclic aromatic hydrocarbons |
| Ph | Phenanthrene |
| Pyr | Pyrene |
| RDS | Recovery determination standard |
| SDL | Sample detection limit |
| SIM | Single ion monitoring |
| SRM | Standard reference material |
| STD | Standard deviation |
5. Acknowledgments
The authors wish to thank the Biosciences Workshop for building and maintaining the atmospheric samplers. The authors thank the MSc and MPhil students from the University of Birmingham who participated in the sampling campaigns. Research described in this article was conducted under contract to the Health Effects Institute (HEI), an organization jointly funded by the United States Environmental Protection Agency (EPA) (Assistance Award No. R-82811201) and certain motor vehicle and engine manufacturers. The contents of this article do not necessarily reflect the views of HEI, or its sponsors, nor do they necessarily reflect the views and policies of the EPA or motor vehicle and engine manufacturers.6. References
- IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Complete List of Agents evaluated and their classification, IARC, 2006 Search PubMed.
- L. Zhu, H. Lu, S. Chen and T. Amagai, J. Hazard. Mater., 2009, 162, 1165 CrossRef CAS.
- R. M. Harrison, D. J. T. Smith and L. Luhana, Environ. Sci. Technol., 1996, 30, 825–832 CrossRef CAS.
- M. Tsapakis and E. G. Stephanou, Environ. Pollut., 2005, 133, 147–156 CrossRef CAS.
- M. Dimashki, L. H. Lim, R. M. Harrison and S. Harrad, Environ. Sci. Technol., 2001, 35, 2264–2267 CrossRef CAS.
- R. Kamens, J. Odum and Z.-H. Fan, Environ. Sci. Technol., 1995, 29, 43–50 CrossRef CAS.
- A. Eiguren-Fernandez and A. H. Miguel, Polycyclic Aromat. Compd., 2003, 23, 193–205 Search PubMed.
- T. Ohura, T. Amagai, M. Fusaya and H. Matsushita, Environ. Sci. Technol., 2004, 38, 77–83 CrossRef CAS.
- L. Z. Zhu and J. Wang, Chemosphere, 2003, 50, 611–618 CrossRef CAS.
- J. Gil-Moltó, M. Varea, N. Galindo and J. Crespo, J. Chromatogr., A, 2009, 1216, 1285 CrossRef CAS.
- J. M. Delgado-Saborit, N. Aquilina, C. Meddings, S. Baker and R. M. Harrison, Environ. Sci. Technol., 2009, 43, 4582–4588 CrossRef CAS.
- R. M. Harrison, J. M. Delgado-Saborit, S. J. Baker, N. Aquilina, C. Meddings, S. Harrad, I. Matthews, S. Vardoulakis and R. Anderson, Measurement and Modeling of Exposure to Selected Air Toxics for Health Effects Studies and Verification by Biomarkers, HEI Research Report 143, Health Effects Institute, Boston, 2009 Search PubMed.
- L. B. Liu, Y. Liu, J. M. Lin, N. Tang, K. Hayakawa and T. Maeda, J. Environ. Sci., 2007, 19, 1–11 Search PubMed.
- L. Bonfanti, M. Careri, A. Mangia, P. Manini and M. Maspero, 19th International Symposium on Column Liquid Chromatography, Innsbruck, Austria, 1995 Search PubMed.
- H. K. Lee, J. Chromatogr., A, 1995, 710, 79–92 CrossRef CAS.
- B. S. Crimmins and J. E. Baker, Atmos. Environ., 2006, 40, 6764 CrossRef CAS.
- EPA, Compendium of methods for the determination of toxic organic compounds in ambient air, US EPA Center for Environmental Research Information, Office of Research and Development, Cincinatti, 1999 Search PubMed.
- Iadn, The IADN team, Version 1.4 – April 2007 edn, 2007, vol. 2009 Search PubMed.
- CARB, California Environmental Protection Agency, Air Resources Board, 3.3 edn, 1998, vol. 2009 Search PubMed.
- M. Shimmo, H. Adler, T. Hyotylainen, K. Hartonen, M. Kulmala and M. L. Riekkola, Atmos. Environ., 2002, 36, 2985–2995 CrossRef CAS.
- A. Cecinato, R. Mabilia and F. Marino, Atmos. Environ., 2000, 34, 5061–5066 CrossRef CAS.
- S. S. Park, Y. J. Kim and C. H. Kang, Atmos. Environ., 2002, 36, 2917–2924 CrossRef CAS.
- S. C. Lee, K. F. Ho, L. Y. Chan, B. Zielinskam and J. C. Chow, Atmos. Environ., 2001, 35, 5949–5960 CrossRef CAS.
- S. P. Wu, S. Tao and W. X. Liu, Chemosphere, 2006, 62, 357–367 CrossRef CAS.
- M. Shimmo, P. Anttila, K. Hartonen, T. Hyotylainen, J. Paatero, M. Kulmala and M. L. Riekkola, J. Chromatogr., A, 2004, 1022, 151–159 CrossRef CAS.
- J. A. Koziel, M. Odziemkowski and J. Pawliszyn, Anal. Chem., 2001, 73, 47–54 CrossRef CAS.
- A. D. St-Amand, P. M. Mayer and J. M. Blais, Atmos. Environ., 2008, 42, 2948 CrossRef CAS.
- A. Albinet, E. Leoz-Garziandia, H. Budzinski and E. ViIlenave, Sci. Total Environ., 2007, 384, 280 CrossRef CAS.
- S. Harrad and L. Laurie, J. Environ. Monit., 2005, 7, 722–727 RSC.
- K. Li, M. Landriault, M. Fingas and M. Llompart, J. Hazard. Mater., 2003, 102, 93–104 CrossRef CAS.
- Z. Fiala, A. Vyskocil, V. Krajak, C. Viau, E. Ettlerova, J. Bukac, D. Fialova and S. Emminger, Int. Arch. Occup. Environ. Health, 2001, 74, 411–420 CrossRef CAS.
- M. S. Callen, M. T. de la Cruz, J. M. Lopez, R. Murillo, M. V. Navarro and A. M. Mastral, Water, Air, Soil Pollut., 2008, 190, 271–285 CrossRef CAS.
- Y. Y. Naumova, S. J. Eisenreich, B. J. Turpin, C. P. Weisel, M. T. Morandi, S. D. Colome, L. A. Totten, T. H. Stock, A. M. Winer, S. Alimokhtari, J. Kwon, D. Shendell, J. Jones, S. Maberti and S. J. Wall, Environ. Sci. Technol., 2002, 36, 2552–2559 CrossRef CAS.
- A. Eiguren-Fernandez, A. H. Miguel, J. R. Froines, S. Thurairatnam and E. L. Avol, Aerosol Sci. Technol., 2004, 38, 447–455 CAS.
- P. C. Vasconcellos, D. Zacarias, M. A. F. Pires, C. S. Pool and L. R. F. Carvalho, Atmos. Environ., 2003, 37, 3009 CrossRef CAS.
- C. Escriva, E. Viana, J. C. Molto, Y. Pico and J. Manes, J. Chromatogr., A, 1994, 676, 375–388 CrossRef CAS.
- M. Bates, P. Bruno, M. Caputi, M. Caselli, G. de Gennaro and M. Tutino, Atmos. Environ., 2008, 42, 6144 CrossRef CAS.
- S. Harrad, Summary of analytical methods and associated quality assurance/quality control (QA/QC) procedures for semi-volatile organic compounds, Division of Environmental Health & Risk Management, School of Geography, Earth & Environmental Sciences, University of Birmingham, Birmingham, 2005 Search PubMed.
- Z. Li, A. Sjodin, E. N. Porter, D. G. Patterson Jr., L. L. Needham, S. Lee, A. G. Russell and J. A. Mulholland, Atmos. Environ., 2009, 43, 1043 CrossRef CAS.
- Z. Vecera, A. Bartosikova, J. Sklenska and P. Mikuska, Chromatographia, 2005, 61, 197–200 CrossRef.
- L. H. Lim, PhD thesis, University of Birmingham, 1999.
- S. S. H. Ho and J. Z. Yu, J. Chromatogr., A, 2004, 1059, 121–129 CrossRef CAS.
- S. S. H. Ho, J. Z. Yu, J. C. Chow, B. Zielinska, J. G. Watson, E. H. L. Sit and J. J. Schauer, J. Chromatogr., A, 2008, 1200, 217 CrossRef CAS.
- USEPA, Quality Assurance Handbook for Air Pollution Measurement Systems. Volume II Par 1. Ambient Air Quality Monitoring Program Quality System Development, EPA-454/R-98–004, US EPA, Office of Air Quality, Planning and Standards, Research Triangle Park, 1998 Search PubMed.
- R. M. Harrison, D. J. T. Smith and L. Luhana, Environ. Sci.Technol., 1996, 30, 825–832 CrossRef CAS.
- C. Schauer, R. Niessner and U. Poschl, Environ. Sci. Technol., 2003, 37, 2861–2868 CrossRef CAS.
- J. Ospital, J. Cassmassi and T. Chico, Multiple Air Toxics Exposure Study in the South Coast Air Basin. MATES III Draft Final Report, South Coast Air Quality Management District, Diamond Bar, CA (USA), 2008 Search PubMed.
- E. Menichini, N. Lacovella, F. Monfredini and L. Turrio-Baldassarri, Atmos. Environ., 2007, 41, 9518–9529 CrossRef CAS.
- E. Rockens, J. Dumollin and C. Matheeussen, Adv. Air Pollut., 2000, 8, 699–707 Search PubMed.
- A. M. Caricchia, S. Chiavarini and M. Pezza, Atmos. Environ., 1999, 33, 3731–3738 CrossRef CAS.
- L. H. Lim, R. M. Harrison and S. Harrad, Environ. Sci. Technol., 1999, 33, 3538–3542 CrossRef.
- K. F. Chang, G. C. Fang, J. C. Chen and Y. S. Wu, Environ. Pollut., 2006, 142, 388–396 CrossRef CAS.
- United States Pharmacopeia, The national formulary, XXIII, United States Pharmacopeial Convention Inc., Rockville, MD, 1995 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1: Optimization of extraction method. See DOI: 10.1039/b9ay00157c |
| This journal is © The Royal Society of Chemistry 2010 |