Determination of benzene polycarboxylic acids in atmospheric aerosols and vehicular emissions by liquid chromatography-mass spectrometry†
Mahmoud Mohammad
Yassine
and
Ewa
Dabek-Zlotorzynska
*
Analysis and Air Quality Section, Air Quality Research Division, Atmospheric Science and Technology Directorate, Science and Technology Branch, Environment Canada, 335 River Road, Ottawa, ON K1A 0H3, Canada. E-mail: ewa.dabek@ec.gc.ca; Fax: +613-990-8568
First published on 24th November 2009
Abstract
A simple, sensitive and reliable LC-ESI(−)/MS method was developed for determination of selected aromatic dicarboxylic acids (e.g. phthalic acid isomers) and tricarboxylic acids (e.g. trimellitic, trimesic) in atmospheric aerosols and vehicular emissions without complex sample pre-treatment. Gradient liquid chromatographic separation was performed on a Zorbax SB-Aq (150 × 2.1 mm i.d., 3.5 µm) column using a mobile phase consisting of 0.1% formic acid (eluent A) and methanol (eluent B). The Zorbax SB-Aq column was chosen as it was specifically designed to retain highly polar compounds, allowing the use of highly aqueous mobile phases. The method was demonstrated to be sensitive and precise. In the SIM mode, LODs for all target acids dissolved in double deionized water were in the range of 0.03–0.1 µg/L. The method showed a good inter-day precision of retention time (RSD <0.3%) and peak area (RSD <3%). Satisfactory recoveries, ranging from 89 to 107% for the spiked blanks and extracts of real samples, were obtained. The method has been successfully applied to the quantitative analysis of selected benzene polycarboxylic acids in urban atmospheric aerosols and particulate matter emitted from diesel- and gasoline-powered motors. Preliminary results suggest that 3-hydroxyphthalic acid may be used as a potential tracer for diesel engine emissions.
1 Introduction
Over the last several years, a particularly active research area has been the investigation of the amount and nature of water soluble organic compounds (WSOC) from atmospheric aerosols. The reasons for this are that organic aerosols, and particularly its water-soluble component, may alter the radiative balance of the atmosphere and cause negative impacts on human health.1,2 In general, the WSOC fraction of aerosol is highly complex as it consists of a multitude of individual compounds, which have a variety of sources. These include mono- and dicarboxylic acids, sugar derivatives and other polyfunctional compounds originating from primary emissions, mainly from combustion and biogenic sources, and secondary organic aerosol (SOA) resulting from the reaction of primary volatile organic compounds in the atmosphere.3Among the identified compounds of organic polar particle constituents, aliphatic and benzene polycarboxylic acids seem to be an important compound class due to their possible formation by chemical reaction in atmosphere.4–8 Direct emissions from vehicular exhausts, biomass burning and cooking have also been suggested as their sources.5,9–12 Thus, the presence of these compounds in the ambient air particles is an indication of a mixture from emission sources and atmospheric chemical processes and, therefore, may serve as useful tracers for source apportionment and processes involving organic carbonaceous aerosols when coupled with receptor models. For example, phthalic acid has been proposed as an indicator of SOA; while its isomers (m-phthalic and p-phthalic acids) are reported to be produced dominantly by primary emission from motor vehicles.5,12–14 However, Kawamura and Kaplan9 reported the presence of phthalic acid in PM collected from gasoline and diesel-powered vehicle emission systems, which has been confirmed by our recent studies.15,16 An additional route for formation of phthalic acids and their introduction into the urban atmosphere may involve waste incineration; for example, m-phthalic has been associated with open burning of plastic bags, roadside litter and landfill trash.17 Yet, in aerosol samples containing a mixture of both SOA and primary source emission, the origins of these compounds have not yet been definitely identified. Thus, reliable quantification of these polar compounds can help us to better understand the emissions and atmospheric processes influencing the molecular characteristics of WSOC.
Gas chromatography with mass spectrometry detection (GC-MS) is still the most widely applied technique for the analysis of aerosol organic composition. However, the high polarity and low levels of non-volatile compounds (e.g. acidic organic compounds) preclude direct GC-MS analysis, making their identification and quantification a challenge. As a result, much less information is available on polar oxygenated species presented in water-soluble organic compounds.18,19 Therefore, modern liquid chromatographic and electrophoretic techniques, such as high performance liquid chromatography (HPLC) or capillary electrophoresis (CE), especially coupled to MS have gained interest in the analysis of polar compounds in airborne particulate matter.15,16,20–22 For instance, Anttila et al.23 reported LC-MS methods with use of ion-trap and time-of-flight MS for the determination of organic acids in aerosol samples collected in a coniferous forest. The emphasis of the study was on screening and identification of the compounds (e.g. pinonic acid and pinic acid) that may contribute to the SOA. Another study by Herrmann and co-workers24,25 demonstrated the usefulness of CE/MS techniques for the analysis of particulate phase substituted methoxyphenols and aromatic acids, and for structural elucidation of the SOA components. Our recent study16 reported a CE-ESI(−)/MS method developed for screening and identification of benzene di- and tri-carboxylic acids in atmospheric aerosols and vehicular emission. Additional measurements with flow infusion electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry were used for further structural information acquisition on the unknown compounds (e.g. 3-hydroxyphthalic acid) detected by CE-MS. However, the sensitivity of the CE-MS was not sufficient to quantify the amount of the acids of interest in atmospheric aerosols.
As a result of this limitation, our research presented herein focused on developing a sensitive method by using a LC-MS to obtain quantitative information on the previously identified aromatic carboxylic acids in atmospheric and transportation emitted PM2.5 samples. The study emphasized also on screening and identification of the compounds that may be used as marker compounds from various transportation emission sources and from secondary photochemical processes. Owing to their polar character, aromatic carboxylic acids are well suited for analysis by a reverse-phase LC-MS in the negative mode. In the present work, a Zorbax SB-Aq column with a bonded phase designed to separate polar compounds in highly aqueous mobile phases was used.26
2 Experimental
2.1 Reagents and chemicals
All chemicals used in this study were of the highest purity available, and were used without further purification. HPLC grade methanol (Caledon, Georgetown, ON, Canada) and formic acid (98+% purity; Acros Organics, New Jersey, USA) were used for the preparation of the mobile phase solutions. Double deionized (DDI) water (18 MΩ; Barnstead, Dubuque, IA, USA) was used for the preparation of all solutions. The benzene carboxylic acids namely, phthalic, m-phthalic, p-phthalic, 4-methylphthalic, 3-hydroxyphthalic, trimesic and trimellitic acid (either as free acids or as their sodium salts) were purchased from Sigma-Aldrich (Toronto, ON, Canada). Stock solutions of all acids (1000 µg/mL) were prepared by dissolving appropriate amounts of acids or their sodium salts in DDI water. The mixed standards were prepared by diluting the appropriate stock solutions stored at 4 °C. All diluted standards were prepared daily from the mixed stock standard solutions.2.2 LC-ESI/MS conditions
LC-ESI/MS analyses were performed with an Agilent 1100 series LC system (Agilent Technologies, Wilmington, DE, USA) consisting of a vacuum degasser, quaternary pump, thermostated column compartment, autosampler, and an Agilent 1100 Series single quadrupole mass spectrometer (MS) equipped with a pneumatically assisted electrospray ionization (ESI). The instrument was controlled, and data were acquired and processed using the Agilent ChemStation software (Rev. B.02.01-SR2).For chromatographic separation, a Zorbax SB-Aq column (150 × 2.1 mm i.d., 3.5 µm) with a compatible guard column (12.5 × 2.1 mm) from Agilent Technologies was used. For all experiments, columns were thermostated at 30 ± 0.1 °C. Injection volume of 40 µL was selected after optimization. Gradient elution was carried out using aqueous 0.1% (v/v) formic acid (eluent A) and pure methanol (eluent B) at a flow rate of 0.4 mL/min. The following elution program was used: at the start 97.5% eluent A and 2.5% eluent B; after 1 min the percentage of eluent B was linearly increased to 45% in 26 min. Between runs the column was flushed with 100% eluent B for 3 min (column cleaning) and equilibrated for 10 min using 2.5% eluent B. Prior to use, the eluents were filtered through a 0.22 µm filter with applied vacuum.
Analytes were detected with ESI in negative mode. Nitrogen gas of 99.9% purity, generated from pressurized air by a nitrogen generator (Parker Hannifin Corporation, Tewksbusty, MA, USA) was used as the nebulizer and the desolvation gas. The optimized operating parameters were as follows: desolvation gas flow, 12 L/min; nebulizer pressure, 35 p.s.i. (241 kPa); capillary voltage, −2000 V; fragmentor voltage, 50 V; desolvation temperature, 350 °C. Other operating conditions were as given in the figure captions.
The identification of target compounds was done in full scan mode by matching the retention time and mass spectrum with standards. Final quantification was performed in a selected ion monitoring (SIM) mode for the parent ions of the target acids using external calibration. Calibration curves were generated using a linear regression analysis.
2.3 Samples and sample preparation
All particulate matter (PM2.5 diameter <2.5 µm) samples were collected on 47-mm i.d. Teflon filters. Atmospheric aerosols were collected within the Canadian National Air Pollution Surveillance (NAPS) PM2.5 Speciation monitoring program. The samples were collected using R&P Partisol Model 2300 Speciation samplers operated at a total flow rate of 10.0 L/min over 24 hours. The PM2.5 emitted in the exhaust from diesel (DPM)- and gasoline (GPM)-powered motor vehicles were obtained from the Emission Research and Measurement Section of the Air Quality Research Division, Environment Canada, as part of the Program for Energy Research and Development Project. The analyzed DPM and GPM samples were sampled at a constant flow-rate of 16.7 L/min for 23 min at room temperature. Following sampling, filters were stored in the freezer.The Teflon filter membranes, separated from the polyolefin support ring using a custom-built stainless steel cutter, were each extracted separately in 5 mL pure methanol for 2 hours using a mechanical shaker. All extracts were filtered through a 0.22 µm PTFE filters followed by evaporation to dryness under a gentle stream of dry nitrogen using Reacti-therm™ III Heating module (Pierce; Rockford, IL, USA). The residue was redissolved in 200 µL DDI and sonicated for 20 min using an ultrasonic bath (Branson and SmithKilne, Model Bransonic 42). Blank samples were treated similarly. Finally, the dissolved samples were transferred into LC insertion vials (200 µL, Agilent) for LC-ESI/MS analysis.
3 Results and discussion
3.1 LC-ESI(−)/MS optimization
To establish optimum conditions for LC-ESI(−)/MS separation of the aromatic acids listed in Table 1, a model solution of seven acids at a concentration of 500 µg/L was used. The instrumental and experimental conditions were carefully studied to obtain the best sensitivity, resolution and the shortest analysis time for the acids under study.| Acid | Peak ID | Mr | Molecular formula |
|---|---|---|---|
| 3-hydroxyphthalic | 1 | 182.13 |
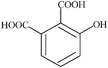
|
| Phthalic | 2 | 166.10 |
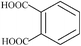
|
| Trimellitic | 3 | 210.14 |
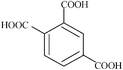
|
| 4-methylphthalic | 4 | 180.10 |
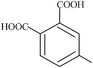
|
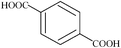
| p-phthalic | 5 | 166.10 |
|
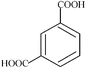
| m-phthalic | 6 | 166.10 |
|
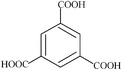
| Trimesic | 7 | 210.14 |
|
A Zorbax SB-Aq column was chosen for this study as it was specifically designed to retain highly polar compounds, such as the studied polycarboxylic acids.26,27 Since the retention of polycarboxylic acids in the reversed phase LC is highly dependent on the degree of ionization of these compounds and, therefore, on the pH of the mobile phase,28,29 acidic mobile phases are preferable. For this purpose, different mobile phase mixtures consisting of 0.1% (v/v) formic acid (pH ∼ 2.7; eluent A) and methanol (eluent B) were tested. Under isocratic conditions, the separation was not good due to the large difference in retention factors (k′ > 30) between the first and the last detected analyte. Thus, a gradient program was optimized in order to get good separation and satisfactory retention of all studied compounds in a reasonable time. The best results were obtained with the gradient profile described in Section 2.2. The isocratic step at the beginning of the run was critical for on-line sample preconcentration to increase the method sensitivity, and thus, lower detection limits. This step allowed injecting up to 40 µL without significant reduction of the peak efficiency. Further increase of the injection volume exceeding 40 µL resulted in severely deteriorating the efficiency as well as the resolution of the separated acids. Using the optimized formic acid-methanol gradient, the LC-MS total ion chromatogram (TIC) demonstrated a baseline separation of model acids in 26 min (Fig. 1).
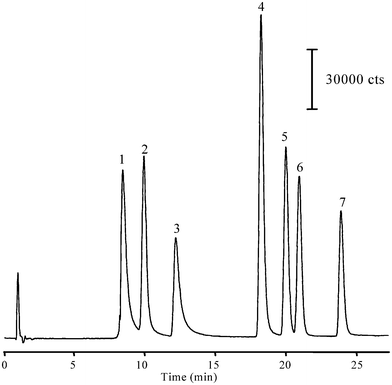 | ||
| Fig. 1 Separation of a model solution of seven benzene polycarboxylic acids at a concentration of 500 µg/L using LC-ESI(−)/MS in SIM mode. LC conditions: Column: Zorbax SB-Aq, 150 × 2.1 mm i.d., 3.5 µm particle size; mobile phase: aqueous 0.1% (v/v) formic acid (eluent A) and methanol (eluent B) delivered at flow rate of 0.4 mL/min; elution profile: isocratic step at 2.5% eluent B for 1 min followed by linear gradient step at a slope of 1.63% eluent B per min for 26 min; injection volume: 40 µL. MS conditions: Drying gas: 12 L/min at 350 °C; Nebulization pressure: 35 psi; Capillary voltage: −2000 V (negative mode); Fragmentor voltage: 50 V; SIM mode programmed to monitor masses: 165, 179, 181, and 209; Peaks: (1) 3-hydroxyphthalic acid; (2) phthalic acid; (3) trimellitic acid; (4) 4-methylphthalic acid; (5) p-phthalic acid; (6) m-phthalic acid; and (7) trimesic acid. | ||
To obtain the best sensitivity and detection, the ESI parameters in negative mode were optimized by running the experiments under the same gradient conditions. The model solution was injected and the detection parameters were optimized. Capillary voltages were checked in the range of −1750 to −3000 V and maximum response was found at −2000 V. Fragmentation voltages were checked in the range 40–70 V and 50 V was found suitable for the target analytes. The desolvation gas flow was evaluated in the range of 6–13 L/min and the maximum response was at 12 L/min at 350 °C. Nebulizer pressure was assessed between 15 and 55 psig (103 and 379 kPa). The highest intensity was achieved at nebulization pressure of 35 p.s.i. (241 kPa). Under these ESI conditions, the studied acids produced intense [M–H]− ion peak with very few fragment ion peaks being observed in the scan mode.
3.2 Analytical performance
Under optimum conditions, the performance and reliability of the proposed method were assessed by determining its analytical figures of merit such as: limit of detection (LOD), limit of quantification (LOQ), linearity, precision and accuracy (as recoveries). In the SIM mode, the LODs based on a signal-to-noise ratio of 3 ranged from 1.2 to 4 pg, which corresponded to LODs ranging from 0.03 to 0.1 µg/L when 40 µL was injected in the column (Table 2). The LOQs based on a signal-to-noise ratio of 10, ranged from 0.1 to 0.3 µg/L (Table 2).| Acid | LOD (µg/L) | LOQ (µg/L) | Intra-day repeatabilitya R.S.D (%) | Inter-day repeatabilityb R.S.D (%) | Correlation coefficientc R2 | ||
|---|---|---|---|---|---|---|---|
| Retention time | Peak area | Retention time | Peak area | ||||
| a n = 10; b n = 3; c Correlation coefficient of the calibration curve. | |||||||
| 3-hydroxyphthalic | 0.07 | 0.23 | 0.08 | 1.6 | 0.30 | 2.8 | 0.9999 |
| Phthalic | 0.06 | 0.20 | 0.07 | 1.4 | 0.20 | 2.4 | 0.9999 |
| Trimellitic | 0.30 | 1.0 | 0.06 | 1.5 | 0.20 | 2.9 | 0.9998 |
| 4-methylphthalic | 0.03 | 0.1 | 0.04 | 1.6 | 0.08 | 2.5 | 0.9999 |
| p-phthalic | 0.07 | 0.23 | 0.03 | 1.3 | 0.13 | 2.5 | 0.9999 |
| m-phthalic | 0.09 | 0.30 | 0.02 | 1.3 | 0.11 | 2.9 | 0.9999 |
| Trimesic | 0.10 | 0.33 | 0.02 | 1.4 | 0.10 | 3.1 | 0.9998 |
The linearity of the method was determined by constructing external calibration curves for seven mixed standards of known concentrations injected in triplicate. The calibration curves were fitted to a linear equation in the range of 0.5–5000 µg/L for all studied acids except for trimellitic acid (1–10000 µg/L). In all cases, the correlation coefficients (R2) were greater than 0.9998 (Table 2). However, the linearity for some of the target analytes, in particular m-phthalic acid (Peak 6) and trimesic acid (Peak 7), deviated at concentration above 5000 µg/L due to the formation of dimmers (Supporting information Fig. S-1†).
Intra-day (within day) and inter-day (between days) precision of retention time and peak area were evaluated at a concentration of 500 µg/L for each analyte over a 3-day period. As expected, retention time variations were usually smaller intra-day than those between days (<0.08%). The within- and between-day precision of the peak area is also good with less than 3% R.S.D. (Table 2). Similar values were obtained at a low concentration of 10 µg/L (data not shown).
The accuracy was evaluated by applying the entire sample preparation procedure to filter blanks spiked with a mixture of acids at known concentration (25 µg/L) and tested in triplicates. As shown in Table 3, recoveries ranged between 91 ± 4 and 110 ± 4%. Accuracy was also evaluated by spiking extracts of analyzed real samples with a 25-µg/L mixed standard. The results of these analyses, shown in Table 3, provided also satisfactory recoveries ranging between 89% and 106%.
| Acid | Blank | PM2.5c | DPMd | GPMe | ||||
|---|---|---|---|---|---|---|---|---|
| Founda (µg/L) | Recoveryb (%) | Found (µg/L) | Recovery (%) | Found (µg/L) | Recovery (%) | Found (µg/L) | Recovery (%) | |
| a values in parenthesis are in ng/filter; b n = 3; c mass of PM2.5 = 525 µg/filter; d mass of DPM = 1785 µg/filter; e mass of GPM = 536 µg/filter. | ||||||||
| 3-hydroxyphthalic | <LOQ | 101 ± 4 | 71.7 (14.3) | 106 | 80.9 (16.2) | 100 | <LOQ | 103 |
| Phthalic | 10.3 (2.10) | 104 ± 6 | 455 (91.1) | 93 | 162 (32.3) | 95 | 125 (25.0) | 98 |
| Trimellitic | <LOQ | 95 ± 5 | 236 (47.2) | 97 | 324 (64.9) | 94 | 60.6 (12.1) | 107 |
| 4-methylphthalic | 1.60 (0.310) | 94 ± 7 | 98.3 (19.7) | 89 | 35.7 (7.20) | 93 | 81.0 (16.2) | 103 |
| p-phthalic | 0.80 (0.170) | 95 ± 4 | 79.4 (15.9) | 92 | 63.8 (12.8) | 95 | 18.2 (3.60) | 102 |
| m-phthalic | 1.40 (0.280) | 92 ± 4 | 27.7 (5.50) | 90 | 60.3 (12.1) | 91 | 97.0 (19.4) | 94 |
| Trimesic | <LOQ | 91 ± 5 | 14.6 (4.20) | 91 | 24.6 (4.90) | 94 | 62.4 (12.50) | 99 |
3.3 Method application
In order to demonstrate the utility of the developed analytical methodology for analysis of real samples, this LC-MS method was applied to the analysis of urban atmospheric aerosols (PM2.5) collected in summer (Windsor, Ontario), and to the analysis of PM2.5 emitted from a heavy duty diesel truck operating on low sulfur (500 ppm) and from a vehicle operating on a winter-grade commercial gasoline. The only sample pretreatment stage involved filtration of methanol extracts through a 0.22-µm pore filter, evaporation to dryness and dissolution in water.Fig. 2 shows typical total ion chromatograms (TICs) for the studied samples. The quantitative data are reported in Table 3. The MS detection was performed using SIM program to monitor the m/z of 165, 179, 181 and 209. The aromatic acids in the samples were identified by comparing the retention times of chromatographic peaks with those of the standard acids as well as by matching the [M–H]− with those of the target acids. Since the resolution of separation is better than 1.5 and the retention times of the target acids are within 0.3% of the retention times of the standard solutions, the possibility of interferences giving false positive results from monitoring only a single ion by MS is greatly reduced. As shown in Fig. 2, peaks corresponding to all the target acids were identified in both atmospheric PM and DPM samples. This is in agreement with the previous results obtained by CE and CE-MS.15,16,30,31 With exception of 3-hydroxyphthalic acid, all target acids were also detected in the GPM sample. These preliminary results suggest that 3-hydroxyphthalic acid (Peak 1) may be used as a potential tracer for diesel engine emissions. Currently, research is being conducted in our laboratory to analyze more samples from different sources in order to examine whether 3-hydroxyphthalic acid can serve as a molecular tracer for the diesel-powered motor emission.
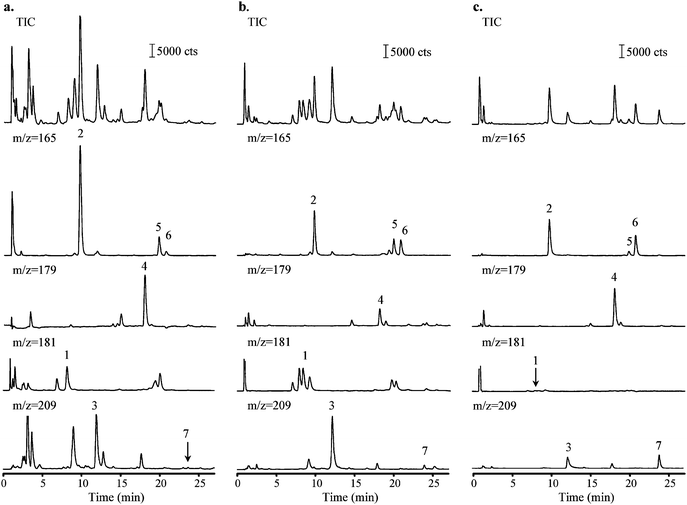 | ||
| Fig. 2 Total and extracted ion chromatograms of (a) urban PM2.5 extract (b) DPM extract and (c) GPM extract. For analytical conditions and peaks identification see Fig. 1 and Table 1. | ||
The samples were also analyzed using MS in scan mode (m/z 100–300). As expected, many other peaks were detected in all samples. Unfortunately, the identities of these peaks could not be determined due to the lack of mass accuracy and MS/MS capability of the quadrupole-MS. Nevertheless, monoterepene oxidation products, in particular pinonic acid and pinic acid, were identified and confirmed in the atmospheric aerosols by matching their retention times with those of standard solutions. Under the optimized conditions, pinonic acid was well separated from the other target acids. However, pinic acid co-elutes with 4-methylphthalic acid (Supporting information Fig. S-2†).
In order to determine if there was any contamination from the Teflon filters and/or sample pre-treatment process, including extraction and filtration, various blanks were determined (Fig. 3). The concentrations of the identified acids in the filter blank are shown in the Table 3. Except for phthalic acid, their levels were near or below LOQ. As expected, the level of the target acids in DDI was comfortably below LODs (Fig. 3a). The extraction solvent (methanol) with and without filtration with PTFE filters was next examined by evaporating 5 mL of pure methanol until dryness and then reconstructing it in 200 µL of water to mimic sample pre-treatment process (Fig. 3b). Blank levels were similar to the filter blank. Comparing with TIC of the unfiltered methanol, no significant difference in the TIC of the filtered methanol was obtained (Fig. 3c). In addition, extract of four blank filters extracted in 5 mL of methanol reveal the same level of concentrations of the target acids (Fig. 3d). These results suggest that the extraction solvent (methanol) is the main source of the contaminants in the Teflon filter blank extract. However, the worst case blank levels for phthalic acid were still only about 2–8% of determined levels in the studied samples.
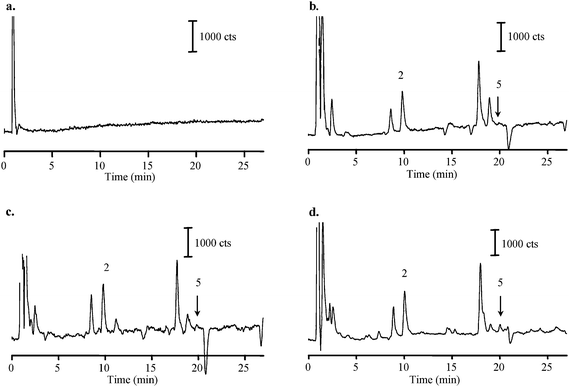 | ||
| Fig. 3 Total ion chromatograms of (a) DDI water; (b) 5-mL of unfiltered HPLC grade methanol evaporated to dryness and redissolved in 200 µL of DDI water; (c) filtered HPLC grade methanol; (d) extract of four Teflon filter blanks. In cases (c) and (d), 5-mL of methanol was filtered using syringe PTFE filters. For analytical conditions and peaks identification see Fig. 1 and Table 1. | ||
4 Conclusions
A new LC-ESI(−)/MS method has been developed and validated for the determination of selected benzene polycarboxylic acids in atmospheric and vehicular emitted PM2.5. The gradient elution profile, using a mobile phase composed of formic acid (0.1%) and methanol, was employed to decrease the time of highly retained species. Seven aromatic polycarboxylic acids were eluted within 26 min. More importantly, the developed LC-ESI(−)/MS method allows a quantification of benzene di- and tri-carboxylic acids with detection limits as low as 0.1 µg/L or better without prior tedious sample preparation. All target acids, including 3-hydroxyphthalic acid, were identified in both atmospheric and diesel emitted PM2.5 samples. Other acids, such as pinonic and pinic (secondary organic aerosol markers from biogenic emission) were also detected in atmospheric PM2.5. However, 3-hydroxyphthalic acid was not detected in a gasoline emitted PM2.5 samples suggesting that 3-hydroxyphthalic acid may be used as a potential tracer for diesel engine emissions. Continuing work in our laboratory is focused on analyzing more samples from different sources in order to examine whether 3-hydroxyphthalic acid can serve as a molecular tracer for the diesel-powered motor emission.Acknowledgements
This work was supported in part by the Canadian Federal Program on Energy Research and Development under PERD 2.1.1. Project “Support the Development of Technological and Other Measures to Control and Reduce Emissions of Particulate Matter”.References
- M. C. Facchini, S. Decesari, M. A. Mircea, S. Fuzzi and G. Loglio, Atmos. Environ., 2000, 34, 4853–4857 CrossRef CAS.
- M. Kanakidou, J. H. Seinfeld, S. N. Pandis, I. Barnes, F. J. Dentener, M. C. Facchini, R. Van Dingenen, B. Ervens, A. Nenes, C. J. Nielsen, E. Swietlicki, J. P. Putaud, Y. Balkanski, S. Fuzzi, J. Horth, G. K. Moortgat, R. Winterhalter, C. E. L. Myhre, K. Tsigaridis, E. Vignati, E. G. Stephanou and J. Wilson, Atmos. Chem. Phys., 2005, 5, 1053–1123 CAS.
- X. Ding, M. Zheng, L. Yu, X. Zhang, R. J. Weber, B. Yan, A. G. Russell, E. S. Edgerton and X. Wang, Environ. Sci. Technol., 2008, 42, 5171–5176 CrossRef CAS.
- W. F. Rogge, M. A. Mazurek, L. M. Hildeman, C. G. Cass and B. R. T. Simoneit, Atmos. Environ., 1993, 27A, 1309–1330 CrossRef CAS.
- P. M. Fine, B. Chakrabarti, M. Krudysz, J. J. Schauer and C. Sioutas, Environ. Sci. Technol., 2004, 38, 1296–1304 CrossRef CAS.
- R. J. Sheesley, J. J. Schauer, E. Bean and D. Kenski, Environ. Sci. Technol., 2004, 38, 6491–6500 CrossRef CAS.
- J. M. Jaeckels, M.-S. Bae and J. J. Schauer, Environ. Sci. Technol., 2007, 41, 5763–5769 CrossRef CAS.
- C. Oliveira, C. Pio, C. Alves, M. Evtyugina, P. Santos, V. Goncalves, T. Nunes, A. J. D. Silvestre, F. Palmgren, P. Wahlin and S. Harrad, Atmos. Environ., 2007, 41, 5555–5570 CrossRef CAS.
- K. Kawamura and I. R. Kaplan, Environ. Sci. Technol., 1987, 21, 105–110 CAS.
- M. El-Fadel and Z. Hashisho, Crit. Rev. Environ. Sci. Technol., 2001, 31, 125–174 CrossRef CAS.
- J. J. Schauer, M. J. Kleeman, G. R. Cass and B. R. T. Simoneit, Environ. Sci. Technol., 2002, 36, 567–575 CrossRef CAS.
- M. P. Fraser, C. G. Cass and B. R. T. Simoneit, Environ. Sci. Technol., 2003, 37, 446–453 CrossRef CAS.
- M. Jang and S. R. McDow, Environ. Sci. Technol., 1997, 31, 1046–1053 CrossRef CAS.
- J. Ray and S. R. McDow, Atmos. Environ., 2005, 39, 7906 CrossRef CAS.
- E. Dabek-Zlotorzynska and M. Piechowski, Electrophoresis, 2007, 28, 3526–3534 CrossRef.
- M. M. Yassine, E. Dabek-Zlotorzynska and P. Schmitt-Kopplin, Electrophoresis, 2009, 30, 1756–1765 CrossRef CAS.
- B. R. T. Simoneit, P. M. Medeiros and B. M. Didyk, Environ. Sci. Technol., 2005, 39, 6961–6970 CrossRef CAS.
- P. Saxena and L. N. M. Hildemann, J. Atmos. Chem., 1996, 24, 57–109 CAS.
- E. G. Stephanou, Nature, 2005, 434, 31–31 CrossRef CAS.
- J. Pol, B. Hohnova, M. Jussila and T. Hyotylainen, J. Chromatogr., A, 2006, 1130, 64–71 CrossRef CAS.
- J. D. Surratt, S. M. Murphy, J. H. Kroll, N. L. Ng, L. Hildebrandt, A. Sorooshian, R. Szmigielski, R. Vermeylen, W. Maenhaut, M. Claeys, R. C. Flagen and J. H. Seinfeld, J. Phys. Chem. A, 2006, 110, 9665–9690 CrossRef CAS.
- R. Winterhalter, M. Kippenberger, J. Williams, E. Fries, K. Sieg and G. K. Moortgat, Atmos. Chem. Phys., 2009, 9, 2097–2117 CAS.
- P. Anttila, T. Hyotylainen, A. Heikkila, M. Jussila, J. Fineli, M. Kulmala and M.-L. Riekkola, J. Sep. Sci., 2005, 28, 337–346 CrossRef CAS.
- C. Müller, Y. Iinuma, O. Böge and H. Herrmann, Electrophoresis, 2007, 28, 1364–1370 CrossRef CAS.
- Y. Iinuma and H. Herrmann, J. Chromatogr., A, 2003, 1018, 105–115 CrossRef CAS.
- http://www.chem.agilent.com/ .
- R. E. Majors and M. Przybyciel, LC.GC Eur., 2002, 2–7.
- U. D. Neue, C. H. Phoebe, K. Tran, C.Y.-F. and Z. Lu, J. Chromatogr., A, 2001, 925, 49–67 CrossRef CAS.
- T. J. Whelan, M. J. Gray, P. A. Slonecker, R. A. Shalliker and M. A. Wilson, J. Chromatogr., A, 2005, 1097, 148–156 CrossRef CAS.
- E. Dabek-Zlotorzynska, R. Aranda-Rodriguez and L. Graham, J. Sep. Sci., 2005, 28, 1520–1528 CrossRef CAS.
- J. Rudolph and J. Stupak, J. Chromatogr. Sci., 2002, 40, 207–213 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S-1 and Fig. S-2. See DOI: 10.1039/b9ay00106a |
| This journal is © The Royal Society of Chemistry 2010 |