Effect of diluent chain length on the performance of the electrochemical DNA sensor at elevated temperature†
Weiwei
Yang
and
Rebecca Y.
Lai
*
Department of Chemistry, University of Nebraska-Lincoln, Lincoln, NE 68588, USA. E-mail: rlai2@unl.edu; Fax: +1 402 472 9402; Tel: +1 402 472 5340
First published on 6th October 2010
Abstract
Here we report the effect of passivating diluent chain length and sensor interrogation temperature on the electrochemical DNA (E-DNA) sensor's mismatch discrimination capability. Both stem-loop and linear probe-based E-DNA sensors were constructed with various diluents, including 6-mercapto-1-hexanol and longer chain hydroxyl-terminated alkanethiols. Contrary to previously reported results, we find that the E-DNA sensors work optimally in the presence of the longer chain diluents, signified by the enhanced % signal suppression observed upon target hybridization. Of note, the sensors' signaling efficiency maintains even when interrogated at an elevated temperature, permitting the use of stringent temperature conditions to improve sensor specificity. For example, a stem-loop E-DNA sensor fabricated with 8-mercapto-1-octanol, when employed at 47 °C, produces signal suppression of 79%, 35% and 1.6% for the perfect match, single-base mismatch, and 2-base mismatch DNA targets, respectively. In addition to the significant enhancement in sensor discrimination capacity, high temperature operation also improves hybridization kinetics. Our results also suggest that the stem-loop E-DNA sensors demonstrate better mismatch discrimination capability when compared to the linear probe system under the same experimental condition.
Introduction
There are growing demands for point-of-care medical diagnostics and rapid methods for the specific detection of nucleic acids (i.e., DNA and RNA) in a variety of complex samples. As a result, a number of DNA sensing methods have been described in the literature, many of which feature impressive sensitivity and generalizability.1–4 However, the vast majority of these approaches requires the addition of exogenous reagents or utilizes changes in relatively nonspecific properties, such as mass, charge, or electron density, to generate an observable signal.2,5 This, unfortunately, makes many of these approaches time-consuming, reagent-intensive, or sensitive to false positive results.5 Owing to the low electroactive background typically observed in clinical samples, and the promising response speed, cost and convenience of microelectronics,6,7electrochemical methods for DNA detection have attracted increasing attention in recent years.8–13 At least a dozen electrochemical DNA sensing approaches have been developed to date, specifically; the Plaxco group and others have developed a label-free, reusable, near real-time electrochemical DNA (E-DNA) sensing platform.14–17 As an electrochemical equivalent of the optical “molecular beacon” detection system, the E-DNA sensor is comprised of a redox-labeled stem-loop or linear DNA probe covalently attached to an interrogating electrode. In the absence of the target DNA, efficient electron transfer is observed between the probe DNA and the interrogating electrode. Hybridization to the target DNA segregates the redox label from the electrode, impeding collision electron transfer, thus producing a large, readily detectable reduction in the redox current.14,17 This sensing strategy has been proven to be highly sensitive and selective, allowing the sensor to be employed directly in whole blood and other realistically complex biological samples.18–20Excellent mismatch discrimination capability is routinely observed in optical molecular beacons,21,22 suggesting that the E-DNA sensor, its electrochemical analog, should display comparable specificity. Nevertheless, as previously reported, the specificity of the original E-DNA sensor is at best modest. While large signal discrimination was evident between the full complement target and targets with multiple mismatches, a mere 1–5% signal discrimination was observed between the full complement target and the single-base mismatch target.18 This level of specificity is sufficient in discriminating full complement and mismatch target DNA under ideal laboratory conditions, more robust signal discrimination is sought after if this sensor is to be employed in field applications. While there are various ways to improve the sensor's specificity, none have been reported to date without significantly altering the sensor construct and signaling strategy.23–25
Thus motivated, here we utilize temperature stringency and surface modification to enhance sensor signal discrimination between the full complement and mismatch target DNA for both the stem-loop and linear-based E-DNA sensors. In particular, we systematically vary the passivating diluents and explore the effects on sensor's performances, which include signaling efficiency, hybridization kinetics and sensor discrimination capacity for mismatch targets. This combined strategy can undoubtedly expand the applicability of the E-DNA sensor point-of-care medical diagnostics.
Experimental
Materials
A thiol and methylene blue (MB)-modified stem-loop or linear oligonucleotide complementary to the K-rasgene was used as probe DNA (Biosearch Technologies, Inc. Novato, CA). The stem-loop probe (SLP) or linear probe (LP) was modified at the 5′-terminus with a C6-disulfide (HO–(CH2)6–S–S–(CH2)6–5′DNA) linker and at the 3′-end with a MB as redox moiety conjugation (Fig. S1, see ESI†). Target DNA sequences including a perfect match (PM), 1-base mismatch (1-MM) and 2-base mismatch (2-MM) targets obtained via commercial synthesis (PAGE purification, Integrated DNA Technologies, Coralville, IA) were used as received. The sequence information of the probes and targets are shown in the following, the target binding region of the probe is italicized and the mismatches in the targets are in bold.SLP : 5′-HS–(CH2)6–CCGTTACGCCACCAGCTCCAAACGG–C7–NH-MB-3′
LP : 5′-HS–(CH2)6–CCGTTACGCCACCAGCTCCAATGCC–C7–NH–MB-3′
PM Target: 5′-TTG GAG CTG GTG GCG TA-3′
1-MM Target: 5′-TTG GAG CTC GTG GCG TA-3′
2-MM Target: 5′-TTG GAG CTC GTG GCC TA-3′
The reagents 6-mercapto-1-hexanol (C6), 8-mercapto-1-octanol (C8), 9-mercapto-1-nonanol (C9), tris-(2-carboxyethyl) phosphine hydrochloride (TCEP) and trizma base were used as received (Sigma-Adrich, St Louis, MO). All other chemicals were of analytical grade. All the solutions were made with deionized water (DI) purified through a Millipore system (18.2 MΩ cm, Millipore, Billerica, MA). Physiological buffer solution (Phys2 buffer, pH 7.4) consisted of 20 mM Tris, 140 mM NaCl, 5 mM KCl, 1 mM MgCl2 and 1 mM CaCl2.
E-DNA sensor fabrication
Prior to sensor fabrication, gold disk electrodes with geometric area of 0.0314 cm2 (CH Instruments, Austin, TX) were polished with a 0.1 µm diamond slurry (Buehler, Lake Bluff, IL), rinsed with deionized water and sonicated in a low power sonicator to remove bound particulates. They were then electrochemically cleaned by a series of oxidation and reduction cycles in 0.5 M H2SO4. The real surface area of each electrode was estimated based on the amount of charge consumed during the reduction of the gold surface oxide monolayer in 0.05 M H2SO4 and a reported value of 400 µC cm−2 was used for the calculation.26 For the fabrication of E-DNA sensors passivated with C6, C8 and C9, a mixture of 1 µL of 200 µM SLP or LPDNA with 1 µL of 10 mM TCEP was first incubated for 1 h to reduce the disulfide bond of the probe DNA, followed by diluting the solution to 100 µL with PBS (10 mM phosphate buffer, 0.1 M NaCl, pH 7.4). The probe DNA was immobilized onto the surface of gold electrodes by incubating the clean electrodes in the diluted probe DNA solution (2.0 µM) for 1 h. The electrodes were then rinsed with DI water and subsequently passivated with 2 mM of C6, C8 or C9 in H2O for 2 h to displace nonspecifically bound oligonucleotides.Electrochemical measurements
All the E-DNA sensors were analyzed using alternating current voltammetry (ACV) with a frequency of 10 Hz and an amplitude of 25 mV AC potential (CHI 1040A, CH Instruments, Austin, TX). The electrolytes employed were Phys2 buffer. DNA probe-modified gold disk electrodes were used as working electrodes. A platinum wire electrode was used as the counter electrode and a Ag/AgCl (3.0 M KCl) electrode served as the reference electrode. For experiments conducted at an elevated temperature (47 ± 1 °C), a ThermoKool heating block (Barnstead International, Dubuque, IA) was utilized. An anodized aluminium block with custom drilled holes was used as the holder for the glass electrochemical cell to allow high temperature in situ hybridization experiments. Prior to electrochemical interrogation, the electrochemical cell was allowed to equilibrate at the appropriate temperature in electrolyte (Phys2 buffer) for at least 30 min to ensure temperature uniformity. The E-DNA sensor response was measured by incubating the electrodes in various target DNA. The sensors were interrogated at different intervals in the target solution until a stable peak current was obtained. The ratio between the stabilized peak current in the target DNA solution and the peak current in the target DNA-free solution was used to calculate the % signal suppression (SS) caused by the target. Sensor regeneration was achieved by 30 seconds rinsing with DI water.The density of electroactive DNA probes on the electrode surface, Γ*, was determined by integration of charges under the reduction peaks of cyclic voltammetry (CV) at low scan rates (eqn (1)).27
| Γ* = Q/nFA | (1) |
| log ks = αlog (1 − α) + (1 − α)log α − log (RT/nFv) − α(1 − α)nFΔEp/2.3RT | (2) |
Results and discussion
Effect of diluent chain length on E-DNA sensor performance
In this study, the E-DNA sensors were passivated with alkanethiols of various chain lengths, including C6, C8, and C9. Fig. 1 shows the effect of the various diluents on both SLP and LP E-DNA sensors' performance as evaluated by ACV. Independent of the diluent chain length, each sensor exhibits a sharp, well-defined peak around −0.27 V (vs.Ag/AgCl), consistent with the electrochemical reduction of MB under similar experimental conditions. The probe density of these sensors systematically decreases with increasing diluent chain length (Table 1). The sensors, however, response differently to WT-Gly, the PM target. The % signal suppression (SS) observed with the SLP sensors increase from 72% to 93% when the length of diluent is extended from C6 to C9, presumably because in the presence of the longer diluent the probe–target duplex DNA is more sterically constricted and thus impedes efficient electron transfer to the electrode, in particular, when compared to sensors passivated with shorter diluents. The LP E-DNA sensors constructed with the three diluents show similar effect as the SLP sensors, with the exception that the C9-passivated sensor exhibits the lowest % SS. The C9-passivated sensor also shows a reproducibly low MB peak current, suggesting a low probe density which is often linked to poorer % SS upon hybridization.29 Despite minor observable differences between the LP and SLP sensors, all sensors can be regenerated by a 30 s room temperature DI water rinse. The hybridization–regeneration plots (Fig. S2, see ESI†) show that nearly all sensors, except the C9-passivated LP sensor, have excellent regenerability (>90%) and reproducibility. The poor sensor regenerability and reproducibility for C9 LP sensor observed could be attributed to low probe density which is often associated with sensor instability.29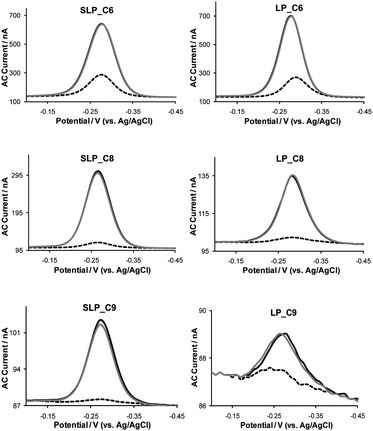 | ||
| Fig. 1 Effect of diluent chain length on E-DNA sensors' signaling efficiency and sensor regeneration capacity in a Phys2 buffer (pH 7.4). Left: SLP sensors; right: LP sensors. Black line: equilibration; dashed line: interrogation with 1.0 µM PM target DNA; grey line: regeneration. | ||
| SLP | LP | |||||
|---|---|---|---|---|---|---|
| C6 | C8 | C9 | C6 | C8 | C9 | |
| a Data are averages of three independent experiments. b Signal suppression is calculated after the sensors have reached signal saturation when challenged with 1.0 µM PM target DNA in a Phys2 buffer. c k s and ks′ are the electron transfer rate constant before and after hybridization with the target DNA, respectively. | ||||||
| Γ*a × 10−12 (molecules cm−2) | 3.1 | 1.3 | 0.2 | 3.2 | 0.76 | 0.16 |
| Signal suppressionb (%) | 72 | 90 | 93 | 78 | 88 | 69 |
| k s c (s−1) | 339 | 185 | 61 | 37 | 5.9 | 1.6 |
| k s′ (s−1) | 239 | –– | –– | 6.3 | 0.70 | 0.13 |
In addition to enhancing signaling efficiency, lengthening of the passivating diluents also improves the sensor's hybridization kinetics (Fig. 2). For example, SLP sensors passivated with C6 show signal saturation within 80 min when challenged with the PM target, whereas SLP sensors fabricated with C8 and C9 display signal saturation time (i.e., time in which ≤0.5% change in the % signal suppression is observed between two data points collected at an interval of 5 minutes) of 40 and 35 min, respectively. This could be explained by the differences in probe density (Table 1), which is generally lower when longer diluents are utilized and low probe density is a leading cause of fast hybridization kinetics.29 However, we also notice that the capacitance current recorded in CV gradually reduces with increasing diluent chain length (data not shown), suggesting a decrease in the electric double layer, which has been shown to speed up diffusion of the negatively charged target DNA to the electrode surface, thus shortening the hybridization time.30–32 Despite the systematically shorter hybridization time due to the lack of the duplex stem structure evident in LP sensors, LP sensors exhibit the same enhancement in hybridization kinetics when longer chain diluents are employed.
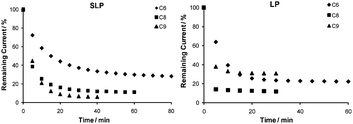 | ||
| Fig. 2 The length of the passivating diluents significantly alters the E-DNA sensors' hybridization kinetics. Shown are the remaining signal (%) versus hybridization time of the sensors when challenged with 1.0 µM PM target DNA. Left: SLP sensors and right: LP sensors. | ||
Effect of diluent chain length on electron transfer kinetics
Since the electron transfer rate constant (ks) of the redox label, MB, is highly dependent on its local environment and distance from the electrode, it is thus an important parameter to be evaluated, with the goal of correlating electron transfer kinetics with sensor performance. Using the Laviron equation,28 we calculated the ks of MB before and after hybridization for both SLP and LP E-DNA sensors passivated with various diluents. The ks values obtained for each system are listed in Table 1. In the absence of the target, a ks value of 339 s−1 is obtained for the C6-passivated SLP sensor; however, when C8 and C9 are used, this value reduces to 185 s−1 and 61 s−1, respectively. Since the electron transfer rate constant varies exponentially with the distance, r, between the electrode and the redox moiety platform,33–36 the observed difference in ks values undoubtedly reflects the increase in electronic coupling distance between the MB redox label and the electrode with increasing length of the passivating diluents. The longer chain diluents restrict the probe's flexibility and result in the increase in the average distance between the MB and electrode. In addition, it has also been reported that the electron transfer kinetics is much more sluggish when the electroactive moiety was buried within the monolayer.37Contrary to the SLP E-DNA sensors, the LP sensors display moderately lower ks when identical passivating diluents are utilized (Table 1). This indicates that ks of MB depends largely on the conformation of the probe DNA since both the probes are identical in length and highly similar in sequence. The results here further support that the stem structure in the SLP probe confines MB closer to the electrode surface, thus the average distance between the MB and the electrode is closer than that of the LP for each system. Among the three systems studied, the ks values for the LP sensors passivated with C6, C8 and C9 are 37 s−1, 5.9 s−1 and 1.6 s−1, respectively. The systematic reduction in ks values also suggests the increase in electronic coupling distance between the MB redox label and the electrode with increasing diluent chain length. Of note, the reduction in the ks value among the LP sensors is more prominent than the SLP sensors. For instance, while ks for the C6 LP sensor is about 23 times higher than that of the C9 LP sensor, a mere 5 times decrease in the ks value is observed between the C6 and C9 SLP sensors. These results suggest that the LPprobe is inherently more flexible than the SLP probe; and the increase in the diluents chain length has a more pronounced effect on the electron transfer kinetics of the MB label in the LP sensors compared to the SLP sensors.
To understand the effect of DNA hybridization on electron transfer, we measured the post-hybridization electron transfer rate constant (ks′) for all sensor systems used in this study (Table 1). For the C6-passivated SLP sensor, a ks′ value of 239 s−1, a value rather similar to the pre-hybridization value (339 s−1), is obtained. The similarity between the two values suggests that the remaining AC current originated mostly from the unhybridized SLP probe rather than the hybridized probes that are in the linear duplex state.29 It is likely that the electron transfer rate for the SLP sensor is limited by the intrinsic electron-transfer rate of the redox moiety. The remaining two SLP sensors, however, show ill-defined CV peaks post-hybridization, thus precluding accurate determination of ks′ (data not shown). Unlike the SLP sensors, the LP sensors show a drastic decrease in the ks′ value regardless of the diluents employed. The extent of the reduction appears to increase with increasing diluents chain length. For example, when compared to the pre-hybridization value, the post-hybridization value is reduced by a factor of 6 for the C6-passivated sensor, whereas these values are lowered by a factor of 8 and 12 for the C8 and C9-passivated sensors, respectively. These results suggest that the duplex DNA leads to an increase in the average distance between MB and electrode when compared to their single-stranded conformation, owing to the more rigid structure of double-stranded DNA compared to single-stranded DNA. Furthermore, sensors fabricated with longer diluents display a more pronounced dampening in the electron transfer kinetics, indicating that longer diluents can alter the flexibility differences between the single-stranded DNA probe and probe–target duplex more efficiently.
Effect of diluent chain length on DNA mismatch discrimination
To investigate the effect of diluent chain length on E-DNA sensor's discrimination capacity, we interrogated the sensors with the PM, 1-MM and 2-MM target DNA. The % SS observed with each E-DNA sensor upon hybridization to 1.0 µM of the three target DNA at room temperature (RT, 21 ± 1 °C) is shown in Table 2. We find that when challenged at room temperature all sensors, regardless of the sensor construct and passivating diluents, exhibit only negligible discrimination between the PM and 1-MM targets. These sensors, however, can distinguish between the PM and 2-MM targets.| Signal Suppression (%) | SLP | LP | ||||
|---|---|---|---|---|---|---|
| C6 | C8 | C9 | C6 | C8 | C9 | |
| a Data are averages of three independent experiments. | ||||||
| RT PM | 72 | 90 | 93 | 78 | 88 | 69 |
| RT 1-MM | 67 | 86 | 88 | 74 | 86 | 66 |
| RT 2-MM | 51 | 75 | 76 | 58 | 81 | 66 |
| 47 °C PM | 15 | 79 | 92 | 16 | 83 | 56 |
| 47 °C 1-MM | 3.4 | 35 | 54 | 4.8 | 60 | 45 |
| 47 °C 2-MM | 2.5 | 1.6 | 1.1 | 3.4 | 9.0 | 10 |
High temperatures have been shown to enhance discrimination of DNA mismatches.38–40 Thus motivated, we explored the effect of elevated temperature on the signaling of the E-DNA sensors (Table 2). As expected, we find that discrimination of perfect matched targets against the mismatched ones is improved at elevated temperatures. For example, the PM to 1-MM signal ratio of C6-passivated SLP sensor increases from less than 1.1 at RT to 4.4 at 47 °C, and PM to 2-MM signal ratio rises from 1.4 to 6 at 47 °C (Fig. S3, see ESI†). Despite the apparent improvement in mismatch discrimination, we notice that the total % SS of C6-passivated SLP sensor reduces with increasing temperature. Specifically, we obtain 72% SS upon hybridization to the PM target at RT, whereas a mere 15% SS is observed when challenged with the same target at 47 °C. This effect could be attributed to the enhanced flexibility of the DNA at elevated temperature, allowing efficient electron transfer even in the duplex state. This extent of signaling degradation will undoubtedly limit sensor sensitivity and thus its applicability.
Since the incorporation of longer chain diluents into the SAM has shown to improve signaling efficiency (i.e., % SS upon hybridization) at room temperature, we presume it will produce the same effect on sensor signaling even at elevated temperature. Thus, we fabricated E-DNA sensors with longer chain diluents and investigated their mismatch discrimination capability at 47 °C. As shown in Fig. 3, there is a large disparity in sensor specificity between C8-passivated SLP sensors employed at room temperature and elevated temperature. Specifically, at 47 °C the % SS obtained with the PM target is ∼79%, whereas the 1-MM and 2-MM targets show only 35% and 1.6% SS, respectively (Fig. 3, right). Contrary to the behavior observed at room temperature, the % SS observed for the same targets are 90%, 86% and 75%, respectively (Fig. 3, left). The PM to 1-MM signal ratio increases from 1.0 at RT to 2.3 at 47 °C, and PM to 2-MM signal ratio rises from 1.2 to 49 at 47 °C. Of note, unlike the C6-passivated sensor, elevated temperature does not appear to significantly dampen the signaling efficiency of the C8-passivated sensor. Specifically, 90% SS is observed when the sensor is hybridized to the PM target at RT, while 79% SS is observed with the same target at 47 °C. The lack of degradation in signaling efficiency with sensors passivated with C8 is presumably due to enhanced rigidity of the probe–target duplex induced by the extended alkanethiol diluents.
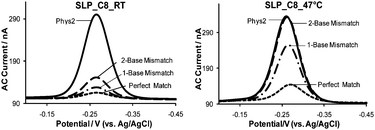 | ||
| Fig. 3 Mismatch discrimination capability of SLP E-DNA sensors passivated with C8 at room temperature (RT, 21 ± 1 °C) and elevated temperature (47 ± 1 °C). The concentration of each target DNA was 1.0 µM in Phys2 buffer (pH 7.4). | ||
In addition to the aforementioned studies in which the sensors were passivated with C8, we have also explored the specificity of sensors fabricated with C9 at 47 °C. The C9-passivated SLP sensor exhibits comparable capability in distinguishing between the PM and 1-MM targets when compared to a C8-passivated sensor. In particular, only 54% SS is observed upon hybridization to the 1-MM target, whereas 92% SS is obtained with the PM target. The C9-passivated SLP sensor can also clearly differentiate between the PM and 2-MM target, in which a mere 1.1% SS is observed in the presence of the 2-MM target. Of note, our results also suggest that the SLP sensors generally exhibit better specificity than LP sensors. Table 2 and Fig. S4 (see ESI†) summarize the effect of sensor interrogation temperature and passivating diluents chain length on sensor specificity. The observed improvement attributed to this combined approach is significant; it allows the E-DNA sensors to be employed in the rapid detection of DNA, and more specifically, the detection of single nucleotide polymorphism which is often required in disease diagnosis.
Conclusions
In this study, we investigated the feasibility of enhancing both the SLP and LP E-DNA sensors' mismatch discrimination capability via temperature stringency and surface modifications. Specifically, we studied the sensors' performance when passivated with longer chain diluents and employed at 47 °C. The sensors have shown to work optimally with longer chain diluents, as indicated by the enhanced % signal suppression observed upon hybridization to the full complement target DNA. The lack of degradation in the sensors' signaling efficiency at elevated temperature allows them to be utilized at temperatures suitable for mismatch discrimination. In addition to the enhanced sensor discrimination capacity, higher sensor interrogation temperature significantly improves hybridization kinetics. Our results also suggest that the SLP sensors show slightly better specificity when compared to the LP sensors under the same experimental condition. Lastly, despite the difference in diluent chain length, most sensors are readily regenerable. The exceptional specificity achieved here, together with the selectivity and reusability typical of the E-DNA sensing platform, suggests that these sensors can potentially be used in the detection of single-nucleotide polymorphism for clinical diagnostics.Acknowledgements
This research was supported by the University of Nebraska-Lincoln and Nebraska EPSCoR (EPS-0701892).References
- W. Vercoutere and M. Akeson, Curr. Opin. Chem. Biol., 2002, 6, 816–822 CrossRef CAS
.
- T. G. Drummond, M. G. Hill and J. K. Barton, Nat. Biotechnol., 2003, 21, 1192–1199 CrossRef CAS
- A. Sassolas, B. D. Leca-Bouvier and L. J. Blum, Chem. Rev., 2008, 108, 109–139 CrossRef CAS
- S. Cosnier and P. Mailley, Analyst, 2008, 133, 984–991 RSC
- M. A. Cooper, Anal. Bioanal. Chem., 2003, 377, 834–842 CrossRef CAS
- H. E. Maes, C. Claeys, R. Mertens, A. Campitelli, C. Van Hoof and J. De Boeck, Adv. Eng. Mater., 2001, 3, 781–787 CrossRef CAS
- R. Bogue, Sens. Rev., 2007, 27, 7–13 CrossRef
- P. He, Y. Xu and Y. Fang, Anal. Lett., 2005, 38, 2597–2623 CrossRef CAS
- K. J. Odenthal and J. J. Gooding, Analyst, 2007, 132, 603–610 RSC
- S. Cagnin, M. Caraballo, C. Guiducci, P. Martini, M. Ross, M. SantaAna, D. Danley, T. West and G. Lanfranchi, Sensors, 2009, 9, 3122–3148 CrossRef CAS
- E. G. Hvastkovs and D. A. Buttry, Analyst, 2010, 135, 1817–1829 RSC
- X. Jin, Y. Fei, L. Zhang, X. Liu, G. Shen and R. Yu, Analyst, 2010, 135, 121–126 RSC
- Y. Bai, J. Li, J. Xu and H. Chen, Analyst, 2010, 135, 1672–1679 RSC
- C. Fan, K. W. Plaxco and A. J. Heeger, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9134–9137 CrossRef CAS
- Y. Mao, C. Luo and Q. Ouyang, Nucleic Acids Res., 2003, 31, e108 CrossRef
- C. E. Immoos, S. J. Lee and M. W. Grinstaff, J. Am. Chem. Soc., 2004, 126, 10814–10815 CrossRef CAS
- F. Ricci, R. Y. Lai and K. W. Plaxco, Chem. Commun., 2007, 3768–3770 RSC
- A. A. Lubin, R. Y. Lai, B. R. Baker, A. J. Heeger and K. W. Plaxco, Anal. Chem., 2006, 78, 5671–5677 CrossRef CAS
- W. Yang, J. Y. Gerasimov and R. Y. Lai, Chem. Commun., 2009, 2902–2904 RSC
- S. J. P. Caňete, W. Yang and R. Y. Lai, Chem. Commun., 2009, 4835–4837 RSC
- X. Fang, X. Liu, S. Schuster and W. Tan, J. Am. Chem. Soc., 1999, 121, 2921–2922 CrossRef CAS
- W. Tan, K. Wang and T. J. Drake, Curr. Opin. Chem. Biol., 2004, 8, 547–553 CrossRef CAS
- S. O. Kelley, E. M. Boon, J. K. Barton, N. M. Jackson and M. G. Hill, Nucleic Acids Res., 1999, 27, 4830–4837 CrossRef CAS
- C. J. Yu, Y. Wan, H. Yowanto, J. Li, C. Tao, M. D. James, C. L. Tan, G. F. Blackburn and T. J. Meade, J. Am. Chem. Soc., 2001, 123, 11155–11161 CrossRef CAS
- J. Chen, J. Zhang, K. Wang, X. Lin, L. Huang and G. Chen, Anal. Chem., 2008, 80, 8028–8034 CrossRef CAS
- H. Angerstein-Kozlowska, B. E. Conway, A. Hamelin and L. Stoicoviciu, J. Electroanal. Chem., 1987, 228, 429–453 CrossRef
-
S. Dong, G. Che and Y. Xie, Chemically Modified Electrodes, Science Press, Beijing, 1st edn, 1995, pp. 123 Search PubMed
- E. Laviron, J. Electroanal. Chem., 1979, 101, 19–28 CrossRef CAS
- F. Ricci, R. Y. Lai, A. J. Heeger, K. W. Plaxco and J. J. Sumner, Langmuir, 2007, 23, 6827–6834 CrossRef CAS
- P. Hildebrandt and D. H. Murgida, Bioelectrochemistry, 2002, 55, 139–143 CrossRef CAS
- S. Rentsch, H. Siegenthaler and G. Papastavrou, Langmuir, 2007, 23, 9083–9091 CrossRef CAS
- F. Ricci, N. Zari, F. Caprio, S. Recine, A. Amine, D. Moscone, G. Palleschi and K. W. Plaxco, Bioelectrochemistry, 2009, 76, 208–213 CrossRef CAS
- L. Guo, J. S. Facci and G. McLendon, J. Phys. Chem., 1995, 99, 8458–8461 CrossRef CAS
- J. N. Richardson, S. R. Peck, L. S. Curtin, L. M. Tender, R. H. Terrill, M. T. Carter, R. W. Murray, G. K. Rowe and S. E. Creager, J. Phys. Chem., 1995, 99, 766–772 CrossRef CAS
- J. F. Smalley, S. W. Feldberg, C. E. D. Chidsey, M. R. Linford, M. D. Newton and Y. Liu, J. Phys. Chem., 1995, 99, 13141–13149 CrossRef CAS
- J. J. Sumner, K. S. Weber, L. A. Hockett and S. E. Creager, J. Phys. Chem. B, 2000, 104, 7449–7454 CrossRef CAS
- J. J. Sumner and S. E. Creager, J. Phys. Chem. B, 2001, 105, 8739–8745 CrossRef CAS
- D. J. Caruana and A. Heller, J. Am. Chem. Soc., 1999, 121, 769–774 CrossRef CAS
- H. Kuhn, V. V. Demidov, J. M. Coull, M. J. Fiandaca, B. D. Gildea and M. D. Frank-Kamenetskii, J. Am. Chem. Soc., 2002, 124, 1097–1103 CrossRef CAS
- G.-U. Flechsig, J. Peter, G. Hartwich, J. Wang and P. Gründler, Langmuir, 2005, 21, 7848–7853 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Structure of the dual-labeled DNA probe used in this study, effect of different chain lengths of alkanethiols on E-DNA sensors' regeneration capability, mismatch discrimination capability of SLP E-DNA sensors passivated with C6 at room and elevated temperatures and mismatch discrimination capability of E-DNA sensors fabricated with different passivating diluents at room and elevated temperature. See DOI: 10.1039/c0an00644k |
| This journal is © The Royal Society of Chemistry 2011 |