Bioconjugated lanthanide luminescent helicates as multilabels for lab-on-a-chip detection of cancer biomarkers†
Vanesa
Fernández-Moreira‡
a,
Bo
Song‡
a,
Venkataragavalu
Sivagnanam
b,
Anne-Sophie
Chauvin
a,
Caroline D. B.
Vandevyver
a,
Martin
Gijs
b,
Ilkka
Hemmilä
c,
Hans-Anton
Lehr
d and
Jean-Claude G.
Bünzli
*ae
aLaboratory of Lanthanide Supramolecular Chemistry (LCSL), École Polytechnique Fédérale de Lausanne, BCH 1402, CH-1015 Lausanne, Switzerland. E-mail: jean-claude.bunzli@epfl.ch; Fax: +41 21 693 9825; Tel: +41 21 693 9821
bLaboratory of Microsystems 2, École Polytechnique Fédérale de Lausanne, Station 17, CH-1015 Lausanne, Switzerland
cPerkinElmer, Wallac Oy, P.O. Box 10, FIN-20101 Turku, Finland
dInstitut Universitaire de Pathologie, Université de Lausanne, Rue du Bugnon 25, 1011 Lausanne, Switzerland
eDepartment of Advanced Materials Chemistry, WCU Research & Development Center, Korea University, Sejong Campus, Jochiwon, Chungnam 339 700, South Korea
First published on 20th November 2009
Abstract
The lanthanide binuclear helicate [Eu2(LC2(CO2H))3] is coupled to avidin to yield a luminescent bioconjugate EuB1 (Q = 9.3%, τ(5D0) = 2.17 ms). MALDI/TOF mass spectrometry confirms the covalent binding of the Eu chelate and UV-visible spectroscopy allows one to determine a luminophore/protein ratio equal to 3.2. Bio-affinity assays involving the recognition of a mucin-like protein expressed on human breast cancer MCF-7 cells by a biotinylated monoclonal antibody 5D10 to which EuB1 is attached via avidin-biotin coupling demonstrate that (i) avidin activity is little affected by the coupling reaction and (ii) detection limits obtained by time-resolved (TR) luminescence with EuB1 and a commercial Eu-avidin conjugate are one order of magnitude lower than those of an organic conjugate (FITC-streptavidin). In the second part of the paper, conditions for growing MCF-7 cells in 100–200 µm wide microchannels engraved in PDMS are established; we demonstrate that EuB1 can be applied as effectively on this lab-on-a-chip device for the detection of tumour-associated antigens as on MCF-7 cells grown in normal culture vials. In order to exploit the versatility of the ligand used for self-assembling [Ln2(LC2(CO2H))3] helicates, which sensitizes the luminescence of both EuIII and TbIII ions, a dual on-chip assay is proposed in which estrogen receptors (ERs) and human epidermal growth factor receptors (Her2/neu) can be simultaneously detected on human breast cancer tissue sections. The Ln helicates are coupled to two secondary antibodies: ERs are visualized by red-emitting EuB4 using goat anti-mouse IgG and Her2/neu receptors by green-emitting TbB5 using goat anti-rabbit IgG. The fact that the assay is more than 6 times faster and requires 5 times less reactants than conventional immunohistochemical assays provides essential advantages over conventional immunohistochemistry for future clinical biomarker detection.
Introduction
The last few years have witnessed the advent of novel, more effective, targeted therapies for cancer treatment. As a consequence, modern pathology labs are increasingly confronted with the need to reliably and efficiently identify relevant therapy targets on cancer tissues as well as other surrogate biomarkers.1 In order to respond to this demand, miniaturized bio-analytical systems are being developed2,3 and applied for the detection of biomarkers in malignant tumours4 and for the follow-up of patients with AIDS.5 We have recently provided proof of concept that lanthanide luminescent bioprobes (LLBs)6,7 combined with ‘lab-on-a-chip’ microfluidics technology, allowed fast detection of tumour biomarkers.8 A commercially available europium chelate was used for this first series of experiments. Over the past three years, we have developed binuclear triple-stranded lanthanide helicates,9 which (i) self-assemble in water at physiological pH and room temperature, (ii) have large thermodynamic stability and kinetic inertness, (iii) possess appropriate photophysical properties (quantum yields up to 20%, lifetimes in the 2–2.5 ms range), (iv) are non-cytotoxic (IC50 > 500 µM), and (v) penetrate several cancerous and non-cancerous cell lines by endocytosis, in order to localize in the endoplasmic reticulum and (vi) allow subsequent imaging by time-resolved (TR) luminescence microscopy.10–15 The helicates which display the best properties are [Ln2(LC2)3] (Ln = Eu, Tb; see Scheme 1) and a versatile method for the quantification of DNA fragments and PCR products has been proposed based on the Eu chelate.16 Since lanthanide double binding tags have definite advantages over monometallic probes17,18 we have now developed a protocol for the bioconjugation of [Ln2(LC2)3] by introducing a carboxylic acid moiety at the end of the solubilizing polyoxyethylene pendant, leading to [Ln2(LC2(CO2H))3] (Ln = Eu, Tb), and subsequently coupling the new helicates to avidin or immunoglobulins. Thus we created lanthanide-containing bioconjugates serving as detection probes for immunocyto- and immunohisto-chemical studies. In the first part of this paper, we describe the synthesis and the photophysical and biochemical properties of these bioconjugates and compare their efficiency for the specific detection of MCF-7 cancerous cells to existing commercially available lanthanide-based and organic luminescent probes. In the second part, this methodology is transferred to microfluidic devices which have the advantage of reduced analysis time and reduced reactant volumes. Since conditions for growing cells in microchannels differ from normal cultures,19 we present an optimization of intervening parameters. Finally, we take advantage of the versatility of the ligand H2LC2(CO2H), which sensitizes the luminescence of both EuIII and TbIII, to develop simultaneous, on-chip detection of two biomarkers on formalin-fixed, paraffin-embedded human breast cancer tissues.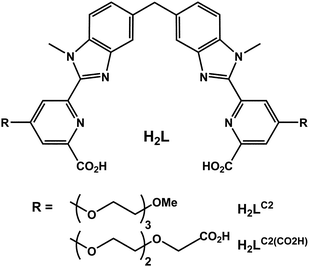 | ||
| Scheme 1 Ditopic hexadentate ligands for the assembly of binuclear lanthanide helicates. | ||
Methodology
We focus herein on the targeting of the human breast adenocarcinoma cell line MCF-7 and on biomarkers expressed by breast carcinomas. We demonstrate that the proposed LLB-on-chip technology reliably detects a mucin-like antigen expressed on MCF-7 cells, as well as estrogen receptor (ER) and human epidermal growth factor receptors 2 (Her2/neu) on formalin-fixed breast cancer tissues. Depending on the targeted biomarker, two types of analyses can be envisaged (Fig. 1). In the immunocytochemical assay (Fig. 1, left panel), a biotinylated monoclonal antibody 5D10 (5D10-B) recognizes its antigen on the cell membrane and subsequently binds to the LLB via strong biotin-avidin coupling (K ≈ 1015 M).20 This helps to increase the signal-to-noise ratio of the luminescence signal in a time-resolved mode. The monoclonal antibody (mAb) 5D10 is one of various mAbs that have been raised against the breast cancer cell line MCF-7,21,22 which is known to express steroid receptors. 5D10 recognizes a mucin-like antigen expressed on most invasive ductal breast carcinomas.23 It has been used as a diagnostic tool24 and found to correlate with the DNA ploidy status of carcinoma cells.25 Targeting the mucin-like antigen26 recognized by 5D10 as a specific marker for MCF-7 cells represents therefore an ideal model system for specific cell imaging with functionalized LLBs.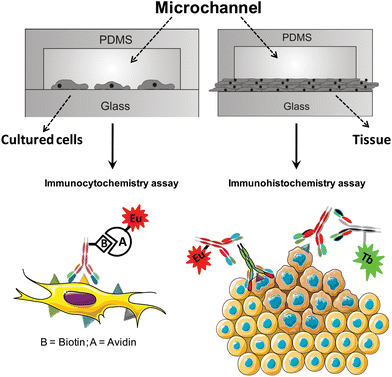 | ||
| Fig. 1 Schematic diagram of the polydimethylsiloxane (PDMS)-glass microfluidic chips used for luminescent imaging of cancer cells seeded in the microchannels (left) and of breast cancer tissues (right). | ||
In the immunohistochemical assay (Fig. 1, right panel), the luminescent lanthanide helicate is bioconjugated to an immunoglobulin, either a goat anti-mouse IgG polyclonal Ab or a goat anti-rabbit IgG Ab. These species-specific secondary antibodies react with either the primary mouse anti-human ER mAb or the rabbit anti-human Her2/neu polyclonal Ab.
Various bioconjugation techniques have been reported to generate LLBs27 and commercial labels are available.28–30 The labelling of a protein by a lanthanide chelate is generally achieved by conjugation between the selected protein and the target agent via a peptide bond and is well documented.31 In our case, the terminal carboxylic function of H2LC2(CO2H) has to be activated, i.e. the –OH group is transformed into an easily leaving group prior to the reaction with the amine moiety.31,32 A common activation pathway is the formation of an N-hydroxysuccinimide (NHS) or sulfo-hydroxysuccinimide (sulfo-NHS) ester.27
Experimental
Starting materials and analytical procedures
Chemicals were purchased from Fluka A.G. and Aldrich and used without further purification unless otherwise stated. Ligand H2LC2(CO2H) was synthesized analogously to H2LC2 or, alternatively, using a new strategic pathway which will be detailed elsewhere.121H NMR (400 MHz, H2O, NaOD 0.05M) δ (ppm): 7.45 (d, J = 2.9 Hz, 2 H, Har), 7.26 (m, 2 H, Har), 7.04 (m, 1 H, Har), 4.23 (m, 2 H, –OCH2–), 3.96 (m, 3 H, –OCH2–and –CH2–), 3.92 (m, 5H, –OCH2– and –NCH3), 3.77 (m, 2 H, –OCH2–), 3.69 (m, 2 H, –OCH2–). C44H42N6O14·1,1 HCl. (found): C, 55.77 (55.74); H, 4.92 (4.91); N, 9.52% (9.49%). The 2:3 complexes were synthesized in situ by mixing stoichiometric quantities of H2LC2(CO2H) and Ln(ClO3)4·xH2O (Ln = Eu, Tb; x = 2.5–4.5) in water at room temperature; the self-assembly process is fast, does not necessitate heating or pH adjustment. The lanthanide salts were prepared from their oxides (Rhône-Poulenc, 99.99%) in the usual way.33 Concentration of the solution was determined by complexometric titrations with a standardized Na2H2EDTA solution in urotropine-buffered medium and with xylenol orange as indicator.34 The Eu-W8044 complex (see ESI†) was obtained from Wallac Oy Turku and used without further purification. It is commercially available from PerkinElmer (product AD0020, Lance Eu-W8044-DTA).MALDI-TOF analyses were carried out on an Axima CFR-Plus instrument (Shimadzu Biotech) equipped with a 337 nm nitrogen laser and operated in positive linear mode. The matrix solutions were prepared with sinapinic acid (20 mg/mL) in CH3CN–H2O–TFA 50:49.9:0.1. For sample preparation, 0.5 mL of the protein solution (0.14 mg/mL) was deposited on the target spot, covered with 0.5 mL of matrix solution and allowed to dry in air. External calibration was carried out with a mixture of standard proteins in the 5–65 kDa mass range. Data interpretation was performed using the Kompact v2.4.3 software. UV-visible spectra were measured in 0.2 cm quartz Suprasil® cuvettes on a PerkinElmer Lambda-900 spectrometer (Table 1). Luminescent spectra and lifetimes were collected on a Horiba-Jobin Yvon Fluorolog FL-3-22 fluorimeter using 0.2 cm quartz Suprasil® cuvettes. All spectra were corrected for the experimental function. Quantum yields were measured by an absolute method using an integration sphere35 and they are given as averages of three independent measurements (Table 1).
| Complex | Absorption | Excitation | Q LLn/% | τ/ms |
|---|---|---|---|---|
| [Eu2(LC2(CO2H))3] | 244 (92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400), 318 (80100), 344 (57900) 400), 318 (80100), 344 (57900) |
322 | 15 ± 2 | 2.45 ± 0.04 |
| EuB1 | 248 (104400), 324.5 (81400), 342 (62700) |
320 | 9.3 ± 0.9 | 2.17 ± 0.01 |
Synthesis of the bioconjugated helicates (Scheme 2)
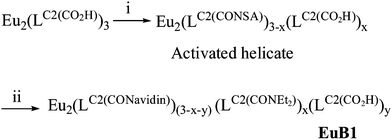 | ||
| Scheme 2 Synthesis of the Eu-labelled avidin conjugate. Synthesis and conditions: (i) EDC, HNSA, DMF(dry), N2, rt (15 min). (ii) (a) Avidin, 0.1 M in PBS + 0.5 M NaCl, pH 8.2, rt (2 h); (b) EtNH2·HCl (10–15 min). | ||
SDS-PAGE electrophoresis
SDS-PAGE electrophoresis was performed using the Mini-PROTEAN® 3 system. Samples and the kaleidoscope pre-stained standards were solubilized in sample buffer (50 mM Tris-HCl pH 6.8, 10% SDS, 2.5% 2-mercaptoethanol, 10% glycerol) and analyzed in 15% polyacrylamide gels using a 5% stacking gel. Staining solution: Coomassie Blue (0.25%) in isopropanol–glacial acetic acid–water (25:10:65); de-staining solution: 7% glacial acetic acid in water. After electrophoresis, pre- and post-stained gels were visualized with a Gene Genius Bio Imaging System using a UV-trans-illuminator and upper white light.Cell lines and culture
The human breast adenocarcinoma cell line MCF-7 (ATCC HTB-22) and the human cervical adenocarcinoma cell line HeLa (ATCC CCL-2) were cultivated in 75 cm2 culture flasks using RPMI 1640 (Sigma, R8758, UK) supplemented with 5% fetal calf serum (FCS), 2 mM L-glutamine, 1 mM sodium pyruvate, 1% non-essential amino-acids, 1% 4-(2-hydroxyethyl)-monosodium salt (HEPES) (all from Gibco® Cell Culture, Invitrogen, Basel, Switzerland). Cultures were maintained at 37 °C under 5% CO2. The growth medium was changed every other day until the time of the experiments. Cell density and viability, defined as the ratio of the number of viable cells over the total number of cells, of the cultures were determined using trypan blue staining and a Neubauer improved hemacytometer (Blau Brand, Wertheim, Germany).ELISAs
Immunoluminescence
MCF-7 and HeLa cells were grown on an 8-well µ-slide (from ibidi, Basel) for 12 h and after washing the slide four times with PBS, the cells were fixed with 0.4% glutaraldehyde for 10 min at rt. They were subsequently washed four times with PBST and treated with 100 µL of blocking buffer for 30 min at rt. The primary antibodies, 5D10-B (10 µg/mL), or α-myco-B, or no mAb for the MCF-7 cells and 5D10-B (10 µg/mL) for HeLa cells, were added to the wells. There were incubated for 1 h at rt. After further washings with PBST (4×), the detection conjugates, EuB1, EuB2, or FiB3 (5 µg/mL), were added to each well and incubated for an additional 1 h at rt. Finally, the slides were washed 4× with PBST and immunoluminescence was measured with a home-modified time-resolved luminescence setup Signifier from Wallac Oy equipped with a Nikon Eclipse 600 microscope and an ORCA-ER CCD camera (Hamamatsu). The following measuring conditions were used for Eu detection: λexc = 340 nm (bandpass filter, BP = 70 nm); λem = 420 nm (longpass LP filter); excitation pulse length, 10 µs; delay time, 100 µs; gate time, 600 µs; exposure time, 60 s. Fluorescence of FiB3 was collected with conventional fluorescence microscopy: λexc = 480 nm (BP filter, 30 nm); λexc = 530 nm (BP filter, 30 nm); exposure time: 10 s.Statistical comparison between the time-resolved luminescent Ln-based and FITC-based detection has been performed as follows: three luminescence/fluorescence images were taken – on different locations of the microslide – and the luminescence/fluorescence intensity of 5 cells/image was calculated using the program AxioVision Rel. 4.5 (Zeiss). The average recorded luminescence/fluorescence intensity was calculated and compared.
Microfluidic devices
Microfluidic channels in PDMS were realized by a replica moulding technique.38 The master structure was made by generating 200 µm wide microfluidic channel patterns in a 100 µm thick SU8 photoresist layer using conventional photolithography. A 10:1 mixture of PDMS pre-polymer and curing agent was cast over the master and then cured at 70 °C for 4 h. Then the cured PDMS replica was peeled-off from the mould and access holes of 0.5 mm diameter for the inlet and outlet tube connections were made by piercing the replica using a blunt needle. The PDMS replica and a glass slide were subjected to air plasma (500 W, 4 mbar) for 20 s and then immediately brought into contact, resulting in permanent bonding. For the breast cancer tissue chips, a tissue section was deposited on a glass substrate, after which the PDMS replicated structure was mechanically clamped to seal the channel.Immunocytochemical assay
Prior to cell loading, microchannels were exposed to UV light for 10 min, in order to prevent bacterial contamination, and were then incubated with 80 µg/mL fibronectin (FN, F1141, Sigma) for 15 min at rt. After washing the coated microchannels with 20 µL of PBS, the appropriate cell solutions were pumped through the microchannels and the device was incubated overnight at 37 °C under 5% CO2. To perform the immunoassay, channels were charged with the required solutions at a flow rate of 50 nL/s and a volume of 2 × 4 µL. The cells grown in the microchannels were fixed with 2% glutaraldehyde for 5 min at rt. After washing with PBS, the microchannels were blocked for 5 min at rt with blocking buffer and incubated with the primary antibody (5D10-B) diluted in blocking buffer (0.5–100 µg/mL) at rt for 5 min. The microchannels were washed with PBST and incubated with 50 µg/mL EuB1, EuB2, or FiB3 diluted in blocking buffer. Finally, the microchannels were washed with PBST and immunostaining was evaluated with luminescence microscopy – see the protocol ‘Immunoluminescence’ above.Immunohistochemical assay
Sections (4 µm thick) of breast carcinomas were obtained from paraffin blocks of formalin-fixed tumour tissues archived in the routine diagnostic laboratory of the Institute of Pathology. All carcinomas were evaluated for hormone receptor and Her2/neu expression by the pathologist involved in this study and sections mounted on Superfrost® gold slides transferred to the LCSL laboratory in a linked-anonymized, coded fashion according to standard procedures.39 The tissues were de-waxed in 100% xylene (10 min) and then rapidly rehydrated successively in 100%, 95%, 70%, and 40% ethanol. After heat-induced antigen retrieval in sodium citrate buffer (S1700, DAKO) using a water bath (95–99 °C, 30 min), the sections were transferred onto the lab-on-a-chip device. The tissues were washed with Tris-HCl buffer pH 7.6 (TBS) and incubated with the primary antibody (non-diluted anti-human ER mouse mAb (RTU-ER-6F11, Novocastra Lab. Ltd.) and a 1/400 dilution of the polyclonal rabbit anti-human c-erbB-2 (Her2/neu) oncoprotein Ab (A 0485, DAKO)) at rt for 8 min. After washing with TBS, the above-described mixture of the two secondary antibodies was added at rt for 8 min: for ER detection, 20 µg/mL EuB4-labelled goat anti-mouse IgG antibody was applied and for Her2/neu detection, we used 20 µg/mL TbB5-labelled goat anti-rabbit IgG antibody. Finally, tissue sections were washed with TBS before TR immunoluminescence detection. The following measurement conditions were used for multiplex detection: EuB4, excitation: 340 nm (BP 70 nm); emission: LP 590 nm; for TbB5: excitation: 340 nm (BP 70 nm); emission: 545 nm (BP 35 nm), other conditions were as described above.Results and discussion
Synthesis and characterization of Eu-labelled avidin
As an initial attempt to conjugate the binuclear lanthanide helicates to avidin, the cross-linker sulfo-NHS and the dehydrating agent EDC were used to activate the carboxylate groups of [Eu2(LC2(CO2H))3].27 The reaction was performed in aqueous solution, using helicate–sulfo-NHS–EDC ratios ranging from 1:6:6 to 1:200:200, reaction times ranging from 15 min to 24 h and pH from 5.6 to 8.2. In each case, however, the coupling yield was found to be too low under any of the tested conditions, so that we turned to water-soluble 1-hydroxy-2-nitrobenzene-4-sulfonic acid (HNSA) as cross-linker agent (Scheme 2).40HNSA-ester derivatives absorb below 300 nm whereas HNSA dianions, which are released upon nucleophilic reactions, give a characteristic absorption band at 406 nm, which allows easy monitoring of the conjugation reaction. The successful bioconjugation process involved two steps. First, the carboxylic acid functions of [Eu2(LC2(CO2H))3] were activated with a 10-fold molar excess of HNSA/EDC during 15 min in dry DMF. Second, the protein dissolved in PBS buffer (pH 8.2) was added to the activated helicate after solvent removal and the evolution of the reaction was monitored by UV-spectroscopy, see Fig. S2 (ESI†). Quenching of the reaction by ethylamine hydrochloride yielded the desired luminescent conjugate EuB1 which presents a statistical repartition of its six arms fitted either with a carboxylate, or with carboxamides (–CONEt or –CONavidin moieties). The coupling conditions were optimized with respect to pH, reaction time, reagent ratios, and reproducibility (see Table S1, ESI†). The yield of the activation step and the targeting ability of the activated helicate are the two factors that need to be optimized in order to have the most favourable number of targeting species at the surface of avidin. The best result regarding the average number of helicates conjugated to avidin fragments, the so-called luminophore/protein ratio (L/P) for EuB1 was achieved with a 10-fold molar excess of the activated helicate at pH 8.2. A considerable improvement of the L/P ratio, from ca. 0.15 to ca. 3.2, was obtained by reducing the activation time from 12 h to 15 min and the coupling time from 48 h to 2 h. Such an increase in L/P can be explained by the ongoing competition between hydrolysis of the activated HNSA-ester and its conjugation to avidin. Moreover, the folding of avidin is pH dependent41 and at pH > 8, the lysine amino groups are deprotonated and might be oriented toward the avidin surface due to the electrostatic repulsion with the lipophilic part of the avidin. As a result, high pH values allow lysine residues to react easily and cleanly with the activated helicate. Further investigation of the reaction conditions showed that the ratio of the reagents also seemed to affect the evolution of the experiment, see Table S1 (ESI†). Thus, a 10-fold molar excess of the activated helicate was found to be the most suitable avidin-to-activated helicate ratio. In fact, it seems to be reasonable to use a 10-fold molar excess of targeting agent considering that avidin bears 36 lysine residues susceptible to react with the activated europium helicate, and, as stated before, a high degree of avidin labelling could negatively affect the biological activity of the protein.
Purification of the bioconjugated europium complex EuB1 was performed by cation exchange chromatography (Fig. S3, ESI†). Two well-differentiated peaks were obtained, at retention times of approx. 5 and 10 min. The first peak corresponds to the unreacted helicate and HNSA, and the second peak to the coupled product. Further identification of EuB1 involved protein electrophoresis (SDS-PAGE) performed in parallel with native avidin and a pre-stained internal standard. Upon UV illumination (Fig. 2a), two luminescent bands appeared for EuB1, which were not observed in the case of the native avidin sample. Staining the gel with Coomassie Blue revealed that the luminescent bands belong to molecules with a molecular weight of ca. ½ avidin and ¼ avidin (Fig. 2b). This implies that tetrameric avidin is split into its subunits during the procedure and that Eu chelates are indeed attached to the fragments of the protein, probably in a random way. If the helicate were only absorbed on the surface of avidin, i.e. by electrostatic forces, it would have been eluted completely from the SDS-PAGE gel. These results are also consistent with a lack of cross-linking of the helicate with two lysine residues belonging to two different subunits.
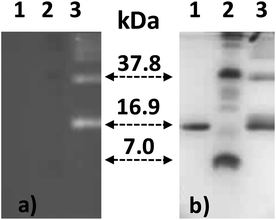 | ||
| Fig. 2 (a) SDS-PAGE gel under luminescence detection, and (b) same gel revealed by staining with Coomassie Blue. Neat avidin (1), pre-stained standard (2), EuB1 (3). | ||
More insight into the products obtained upon bioconjugation was revealed by MALDI-TOF mass spectrometry. For free avidin, four peaks were observed at m/z 62.7, 47.2, 31.5 and 15.7 kDa. These molecular masses correspond to avidin fragments bearing 4, 3, 2, and 1 subunit(s), respectively, dissociation being induced by laser irradiation.42 As expected, all these peaks were shifted towards higher molecular weight after conjugation with the europium helicate (MW 2.9 kDa per complex), as expected. This is exemplified in Fig. 3 which displays two bands corresponding to either a 3-unit fragment coupled to one helicate or to un-fragmented avidin coupled to two helicates. However, no conclusion can be drawn with respect to the labelling efficiency because of the micro-heterogeneity caused by the glycosylation of the avidin polypeptide chains43 as well as a possible dissociation of the luminescent probe upon laser irradiation.
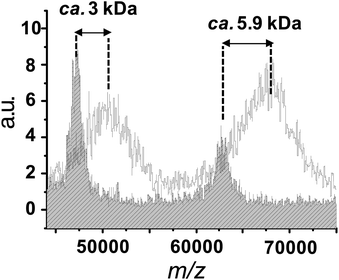 | ||
| Fig. 3 Superposition of the MALDI-TOF mass spectra between 45 kDa and 75 kDa of avidin (patterned spectrum) and EuB1 (plain spectrum). | ||
For this reason, we used UV-visible spectroscopy to reliably calculate the L/P ratio.44 Concentration of the helicate was determined by measuring its absorbance at 320 nm, while the (conjugated) avidin concentration was calculated from its absorption at 280 nm after allowing a correction for the helicate absorption at this wavelength (see Fig. S4, ESI†). In the case of EuB1, L/P was found to be 3.2. Since we used a helicate-to-avidin ratio of 10, the yield of the conjugation reaction is 32%. We have not found comparable data in the literature, but this yield is similar to the one found by Charbonnière and co-workers30 for the labelling of bovine serum albumin (BSA) with activated lanthanide complexes (25%). With such an L/P value, the protein activity is not affected, as subsequently demonstrated by bio-affinity assays (see below).
Luminescence properties of the conjugate EuB1
The functionalized europium complex [Eu2(LC2(CO2H))3] presents a main broad absorption band at 318 nm with a large molar absorption coefficient (log ε324 = 4.91), as well as two shoulders located at 244 and 344 nm (Table 1), similar to the spectrum reported for the parent europium helicate [Eu2(LC2)3] (322, 246, 340 nm).12 In the case of the bioconjugate EuB1, the main absorption band is red shifted by about 6 nm, at 324.5 nm, but the overall shape of the spectrum remains the same. Upon excitation at 320 nm, EuB1 emits the characteristic sharp EuIII lines assigned to the 5D0 → 7FJ transitions (J = 0–5), as shown in Fig. 4 in which the excitation spectrum matches the absorption spectrum. The latter fact demonstrates the sensitization of EuIII luminescence by the benzimidazolepyridine core of the ligands. It is noteworthy that no ligand phosphorescence is recorded.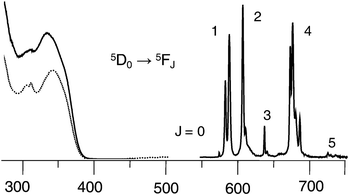 | ||
| Fig. 4 Excitation (—), absorption (⋯), and emission spectra of EuB1 2 × 10−5 M at 295 K in 0.01 M PBS aqueous solution, pH 7.4. | ||
A previous study of EuIII tris complexes with dipicolinic acid derivatives has demonstrated that terminal substituents of polyoxyethylene pendants grafted on the pyridine 4-position have little influence on the coordination sphere and the overall shape of the luminescence spectra. Only a fine tuning of the quantum yield was observed,45 similar to what is observed here. Indeed, the emission spectrum of EuB1 (see details in Tables S2 and S3 in the ESI†) may be interpreted as arising from a main species with a pseudo D3 symmetry as observed for the parent helicate [Eu2(LC2)3].12 The luminescence decays of both [Eu2(LC2(CO2H))3] and EuB1 are single exponential functions corresponding to lifetimes of 2.45 ms and 2.17 ms, respectively, that is an insignificant decrease for the EuB1 conjugate. On the other hand, the quantum yields decrease from 21% for [Eu2(LC2)3]12 to 15% for [Eu2(LC2(CO2H))3], and 9.3% for EuB1. Since the lifetime is relatively unaffected by the conjugation, the drop in quantum yield reflects a less efficient energy transfer, probably due to de-activation of the ligand states by interaction with the protein functionalities. In view of the large absorption coefficient at the excitation wavelength, the overall efficiency of the luminescent conjugate, ε × Q = 7560 M−1 cm−1, remains, however, large enough to provide sufficient detection limit for the purpose of this investigation.
Bio-affinity test and cell imaging
Bio-affinity assays were used to assess the recognition of an antigen by its monoclonal antibody mAb, visualized by a biotin-avidin specific interaction.20,46 Two other luminescent conjugates were used in parallel with EuB1 in order to compare its efficiency: Eu-W8044-labelled avidin (EuB2, see ESI†) and the organic conjugate streptavidin-FITC (FiB3). The first immunoassay was performed using a streptavidin-coated 96-well plate and increasing concentrations of biotinylated rabbit anti-streptavidin Ab (α-SA-B) as primary antibody. Time-resolved detection of the probe luminescence showed that the signal/noise (S/N) ratio for EuB1 relative to EuB2 and FiB3 was 1.2 and 121, respectively. The lowest concentration of α-SA-B giving a detectable signal is 2.5 ng/mL with EuB1, 1 ng/mL with EuB2, and 34 ng/mL in the case of FiB3 (Fig. 5). Thus, bioconjugation of the helicate to avidin in EuB1 does not affect substantially the biological properties of the protein. Furthermore, whereas EuB1 has a similar detection limit and signal/noise ratio as EuB2, the lanthanide probes display much better detection properties than the organic conjugate FiB3.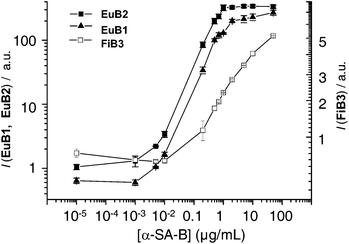 | ||
| Fig. 5 Absolute emission intensity of EuB1 (▲), EuB2 (■) and FiB3 (□) as a function of the biotinylated rabbit anti-streptavidin (α-SA-B) antibody concentration. | ||
In view of these results, indirect immunocytochemistry assays were performed to test the selectivity of EuB1 in comparison with EuB2 and FiB3, using biotinylated 5D10 mAb (5D10-B) as primary antibody. The breast cancer cell line MCF-7 was chosen since this cell line expresses the mucin-like protein26 recognizable by 5D10-B. The specificity of the experiment was monitored with HeLa cells which do not express the antigen recognized by 5D10 and by using the non-specific biotinylated anti-mycoplasma mAb (α-myco-B), see Fig. 6. The three bioprobes displayed selectivity towards MCF-7 cells incubated with 5D10-B. The signal-to-reference (S/R) ratio of each probe was calculated taking the emission intensity from MCF-7/α-myco-B as background value. S/R ratios for EuB1 and EuB2 are comparable (5.8 and 6.8 for EuB1 and EuB2, respectively) and significantly higher than those for FiB3 (1.9). In view of the promising results observed for EuB1, TR luminescence microscopy images were then recorded using exactly the same experimental conditions (Fig. S5, ESI†). As expected from the immunoluminescence results, statistical analysis of the TR luminescence intensity showed a better S/R for the EuIII bioprobes (2.2 and 2.1 for EuB1 and EuB2, respectively) compared to FiB3 measured in conventional mode (1.1), see Fig. 6.
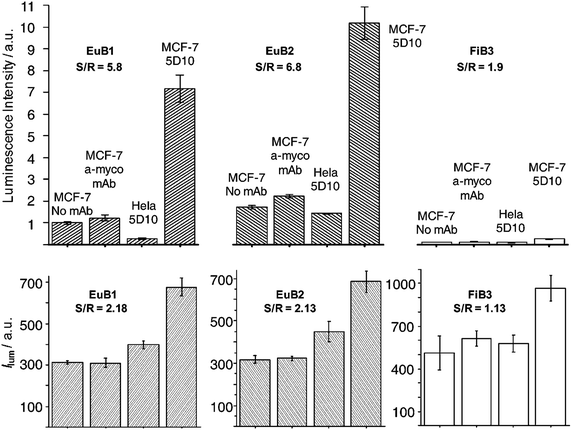 | ||
| Fig. 6 Luminescence intensity of EuB1, EuB2, and FiB3 detection probes as a function of the primary antibody (5D10 vs. α-myco as irrelevant Ab vs. no Ab) and the cell line. Top: immunoluminescence; bottom: statistical analysis of luminescence microscopy images. | ||
Cell culture in microchannels
Prior to cell loading, the microfluidic channels were coated with a cell adhesion protein, poly-L-lysine (PLL) or a major integrin ligand, fibronectin (FN),47 as the PDMS and glass-based microfluidic device does not provide good adhesion conditions for epithelial cells. The choice of the coated cell-adhesion-protein depends on the cell type: MCF-7 cells seeded in FN- and PLL-coated microchannels did adhere and grow in an epithelial monolayer, while this was not the case for uncoated channels; on the other hand, HeLa cells grew into an epithelial monolayer only in FN-coated microchannels (Fig. 7).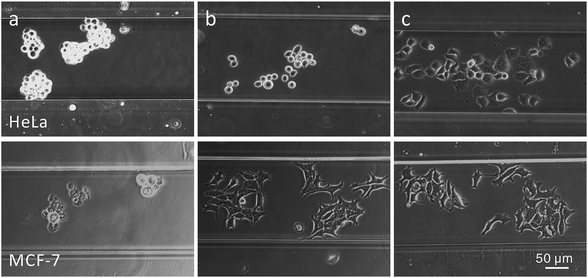 | ||
| Fig. 7 Bright field images of HeLa (upper row) and MCF-7 (lower row) cells seeded during 24 h in (a) uncoated, (b) PLL-coated or (c) FN-coated 200 µm wide microchannels. | ||
The experimental conditions for cultivating cells in the microchannels have been carefully optimized. Firstly, with respect to the concentration of fibronectin (FN): the number of adherent cells (NAD) was found to depend linearly on the FN concentration up to ca. 60 µg/mL and then to level off (Fig. 8a). Based on these pilot experiments, a concentration of 80 µg/mL was chosen for further experiments. Secondly, the coating time of FN was set to 15 min since the NAD was found not to increase with longer coating times (Fig. 8b). Thirdly, the optimal cell seeding time was determined, by injecting a cell solution (1 × 106 cells/mL) into the microchannel and incubating it for times ranging from 0.5 h to 6 h, before excess non-adherent cells were washed out with a phosphate buffer saline (PBS) solution at a flow rate of 100 nL/s. An increase in NAD with increasing cell incubation periods was observed (Fig. 8c). After 0.5 h, MCF-7 cells began to adhere to the surface of the FN-coated channel and then cells spread: many pseudo-pods were formed, the cell fringes enlarged and their area increased. Compared to MCF-7 cells, HeLa cells grew faster and began to adhere and spread as early as 15 min after seeding. After 2 h, increasing the seeding time did not result in increased cell adhesion, but the cells did not have the right morphology, namely an epithelial monolayer, for incubation times of <4 h. Therefore, for all subsequent experiments, a cell seeding time larger than 8 h was adopted, which results in an epithelial monolayer for all cell lines used in this study. Finally, cell seeding density was also examined (Fig. 8d) since highly concentrated solutions of cells can potentially overpopulate and hence prevent proper cell attachment onto the walls of the microchannels. A concentration of 2 × 106 cells/mL provided an adequate number of cells without leading to multilayer growth.
![MCF-7 and HeLa epithelial cell adhesion study in a microchannel. The average number of cells are counted in an area of 2.5 mm2 of the microchannel as a function of (a) FN concentration (FN coating = 2 h, cell seeding time = 8 h, cell density = 1 × 106 cells/mL), (b) FN coating time, ([FN] = 80 µg/mL, same cell seeding time and cell density as for part (a)), (c) cell seeding time ([FN] = 80 µg/mL, FN coating time = 15 min, cell density = 1 × 106 cells/mL), and (d) the cell seeding density ([FN] and FN coating time as in part (c)), seeding time = 8 h.](/image/article/2010/AN/b922124g/b922124g-f8.gif) | ||
| Fig. 8 MCF-7 and HeLa epithelial cell adhesion study in a microchannel. The average number of cells are counted in an area of 2.5 mm2 of the microchannel as a function of (a) FN concentration (FN coating = 2 h, cell seeding time = 8 h, cell density = 1 × 106 cells/mL), (b) FN coating time, ([FN] = 80 µg/mL, same cell seeding time and cell density as for part (a)), (c) cell seeding time ([FN] = 80 µg/mL, FN coating time = 15 min, cell density = 1 × 106 cells/mL), and (d) the cell seeding density ([FN] and FN coating time as in part (c)), seeding time = 8 h. | ||
Immunocytochemical detection of cells in microchannels
To compare our method with standard immunocytochemistry assays, experiments were performed with increasing concentrations of the primary 5D10-B mAb, followed by TR detection of the LLBs or by conventional fluorescence microscopy for FiB3. The lowest concentration of biotinylated 5D10 mAb to give a detectable signal is 0.58 µg/mL with EuB2 and 0.71 µg/mL with EuB1versus 1.4 µg/mL when using FiB3 (Fig. 9).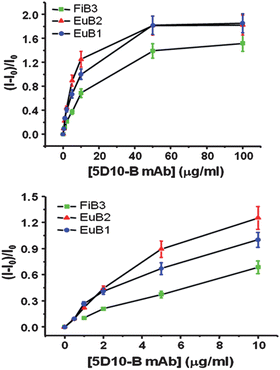 | ||
Fig. 9 On-chip immunocytochemical detection of the mucin-like protein expressed on MCF-7 cells using the monoclonal antibody 5D10. Relative luminescent intensity as a function of 5D10-B concentration using EuB1 ( ), EuB2 ( ), EuB2 ( ) or FiB3 ( ) or FiB3 ( ) as secondary detection probe. ) as secondary detection probe. | ||
The specificity of the assay was monitored as described above. Compared to FITC fluorescence detection, the background luminescence could be completely eliminated in TR detection mode, which avoids the detection of false positive cells. Statistical analysis of the TR luminescence detection evidenced an improvement in the signal-to-noise ratio by a factor of 1.6, compared to classical fluorescence detection (Fig. 10 and Fig. S6, ESI†).
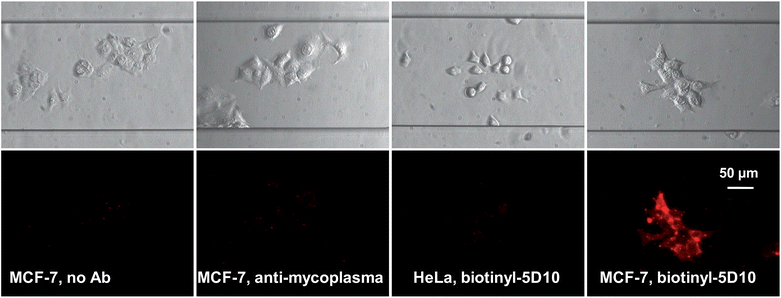 | ||
| Fig. 10 On-chip immunocytochemical detection of the mucin-like protein expressed on MCF-7 cells by 5D10 with the EuB1 bioprobe (Col. 4). Negative controls: no mAb (Col. 1), α-myco-B (Col. 2), and HeLa cells with 5D10-B (Col. 3). | ||
Dual detection of biomarkers on a cancerous tissue
Having demonstrated the feasibility of our approach for the detection of antigens expressed by cells grown in microchannels, we subsequently turned to the analysis of diagnostic biomarkers on human breast cancer tissues. The predictive and prognostic biomarkers for breast cancer, ER and Her2/neu, were detected by an immunohistochemical reaction. Both biomarkers are routinely used in clinical practice, having an established role in predicting tumour response to hormone therapy and to immunotherapy with trastuzumab.48 Simultaneous detection of both tumour markers on a breast cancer tissue in the microfluidic system was tested, taking advantage of the spectral versatility brought by red-emitting Eu-LLB and green-emitting Tb-LLB. However, when performing an indirect immunohistochemical assay on breast cancer tissues, using the biotin-avidin system described above, we were confronted with a high intracellular background signal, which can likely be attributed to the intracellular expression of biotin, often observed in cancer tissues.49 Therefore, it was decided to develop two new LLBs: EuB4, a Eu-labelled goat anti-mouse IgG Ab; and TbB5, a Tb-labelled goat anti-rabbit IgG Ab, as detection antibodies. As shown in Fig. 11, Her2/neu expressed on the cancerous cell surface was detected by TbB5 and ER located within the nuclear membranes of the cancerous cells was evidenced with EuB4. The results were in complete agreement with those obtained in the clinical laboratory using classical immunohistochemistry settings, but were obtained in only 20 min versus the 2 h needed for the classical protocol and with 5 times less reactants.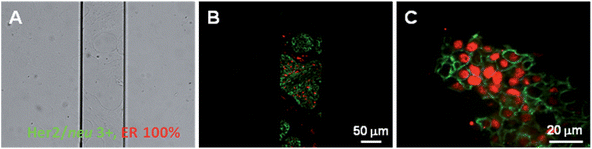 | ||
| Fig. 11 On-chip immunohistochemical detection of Her2/neu and ER in a breast cancer tissue sample. (A) Bright field image; (B) merged luminescent image, Her2/neu detected by green-emitting TbB5 and ER stained with red-emitting EuB4; (C) magnified image. Note the nuclear expression of ER and the strictly membranous decoration of cells with Her2/neu. | ||
Conclusion
We have described herein methodologies for conjugating luminescent lanthanide binuclear helicates to various proteins, as well as for growing epithelial monolayers in microchannels. The new conjugates perform well as detection probes for the targeting via specific recognition of a mucin-like protein expressed on the surface of MCF-7 tumour cells by the 5D10 mAb. The use of time-resolved detection with lanthanide conjugates leads to significantly lower detection limits compared to conventional fluorescence detection via FITC, with markedly improved signal-to-reference (S/R) ratios in immunocytochemical analyses and luminescence microscopy imaging. Similar improvements were obtained with the on-chip microchannel devices.The new helicate conjugates compare well to the mononuclear commercial chelate W8044, with almost identical detection limits and similar S/R or S/N ratios. They present, however, potential advantages: their inherent chirality50 and the possibility of introducing two different lanthanide ions in the same molecular edifice;51 these features are presently being investigated in our laboratories. The real advantage lies in the ditopic ligand system used to self-assemble the helicates and which sensitizes both EuIII and TbIII luminescence. Combining these benefits with microfluidics technology, we were able to realize a novel ultrafast, and economic assay for ER and Her2/neu detection on human breast cancer samples. The improvement in analysis time and the reduction in reagent consumption inherent to this new technology are substantial and with enormous potential for application in the diagnostic routine.
Acknowledgements
This research is supported through grants from the Swiss National Science foundation (200020_119866) and the Swiss Office for Science and Education (C07.0116, within the frame of COST Action D38 from the European Science Foundation). We thank Laure Ménin for the mass spectrometry analysis performed on the complexes and the technical staff of the Institute of Pathology (‘Grand Laboratoire’) for providing sections of tumour tissues. J.-C. B. thanks the World Class University (WCU) program from the National Research Foundation of Korea funded by the Ministry of Education, Science, and Technology (grant Nr. R31-10035).References
- K. Glunde, M. A. Jacobs, A. P. Pathaka, D. Artemov and Z. M. Bhujwalla, NMR Biomed., 2009, 22, 92 CrossRef CAS.
- P. S. Dittrich, K. Tachikawa and A. Manz, Anal. Chem., 2006, 78, 3887 CrossRef CAS.
- T. Laurell, F. Petersson and L. Nilsson, Chem. Soc. Rev., 2007, 36, 492 RSC.
- M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161 CrossRef CAS.
- X. Cheng, A. Gupta, C. Chen, R. G. Tompkins, W. Rodriguez and M. Toner, Lab Chip, 2009, 9, 1357 RSC.
- J.-C. G. Bünzli, Chem. Lett., 2009, 38, 104 CrossRef CAS.
- C. P. Montgomery, B. S. Murray, E. J. New, R. Pal and D. Parker, Acc. Chem. Res., 2009, 42, 925 CrossRef CAS.
- B. Song, V. Sivagnanam, C. D. B. Vandevyver, I. A. Hemmilä, H.-A. Lehr, M. A. M. Gijs and J.-C. G. Bünzli, Analyst, 2009, 134, 1991 RSC.
- J.-C. G. Bünzli, A.-S. Chauvin, C. D. B. Vandevyver, B. Song and S. Comby, Ann. N. Y. Acad. Sci., 2008, 1130, 97 CrossRef CAS.
- C. D. B. Vandevyver, A.-S. Chauvin, S. Comby and J.-C. G. Bünzli, Chem. Commun., 2007, 1716 RSC.
- A.-S. Chauvin, S. Comby, B. Song, C. D. B. Vandevyver and J.-C. G. Bünzli, Chem.–Eur. J., 2007, 13, 9515 CrossRef.
- A.-S. Chauvin, S. Comby, B. Song, C. D. B. Vandevyver and J.-C. G. Bünzli, Chem.–Eur. J., 2008, 14, 1726 CrossRef.
- E. Deiters, B. Song, A.-S. Chauvin, C. D. B. Vandevyver and J.-C. G. Bünzli, New J. Chem., 2008, 32, 1140 RSC.
- B. Song, C. D. B. Vandevyver, A.-S. Chauvin and J.-C. G. Bünzli, Org. Biomol. Chem., 2008, 6, 4125 RSC.
- E. Deiters, B. Song, A.-S. Chauvin, C. Vandevyver and J.-C. G. Bünzli, Chem.–Eur. J., 2009, 15, 885 CrossRef CAS.
- B. Song, C. D. B. Vandevyver, E. Deiters, A.-S. Chauvin, I. A. Hemmilä and J.-C. G. Bünzli, Analyst, 2008, 133, 1749 RSC.
- L. J. Martin, M. J. Hahnke, M. Nitz, J. Wohnert, N. R. Silvaggi, K. N. Allen, H. Schwalbe and B. Imperiali, J. Am. Chem. Soc., 2007, 129, 7106 CrossRef CAS.
- N. R. Silvaggi, L. J. Martin, H. Schwalbe, B. Imperiali and K. N. Allen, J. Am. Chem. Soc., 2007, 129, 7114 CrossRef CAS.
- M. Morel, D. Bartolo, J.-C. Galas, M. Dahan and V. Studer, Lab Chip, 2009, 9, 1011 RSC.
- K. Matsumoto and J. G. Yuan, in Metal Ions in Biological Systems, ed. A. Sigel and H. Sigel, Marcel Dekker Inc., New York, 2003, vol. 40, ch. 6 Search PubMed.
- L. Plessers, E. Bosmans, A. Cox and J. Raus, Anticancer Res., 1985, 5, 609.
- L. Plessers, E. Bosmans, A. Cox and J. Raus, Anticancer Res., 1986, 6, 885 CAS.
- S. C. Brooks, E. R. Locke and H. D. Soule, J. Biol. Chem., 1973, 248, 6251 CAS.
- L. Plessers, E. Bosmans, A. Cox, L. O. Debeeck, J. Vandepitte, J. Vanvuchelen and J. Raus, Anticancer Res., 1990, 10, 271 CAS.
- Y. Chin, L. Plessers, J. Vandepitte and J. Raus, Eur. J. Cancer Clin. Oncol., 1991, 27, 48 CrossRef CAS.
- C. Vandevyver, S. Canarelli, C. Bossen, I. Fisch, K. Motmans, J. Raus and R. Freitag, Biotechnol. Bioeng., 2007, 97, 721 CrossRef CAS.
- G. T. Hermanson, Bioconjugate techniques, Academic Press, Elsevier, Amsterdam, 2008 Search PubMed.
- I. Hemmilä, S. Dakubu, V. M. Mukkala, H. Siitari and T. Lövgren, Anal. Biochem., 1984, 137, 335 CrossRef CAS.
- I. Hemmilä and S. Webb, Drug Discovery Today, 1997, 2, 373 CrossRef CAS.
- N. Weibel, L. J. Charbonnière, M. Guardigli, A. Roda and R. F. Ziessel, J. Am. Chem. Soc., 2004, 126, 4888 CrossRef CAS.
- J. Hovinen and P. M. Guy, Bioconjugate Chem., 2009, 20, 404 CrossRef CAS.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606 RSC.
- J.-C. G. Bünzli, J.-R. Yersin and C. Mabillard, Inorg. Chem., 1982, 21, 1471 CrossRef.
- G. Schwarzenbach, Complexometric Titrations, Chapman & Hall, London, 1957 Search PubMed.
- A. Aebischer, F. Gumy and J.-C. G. Bünzli, Phys. Chem. Chem. Phys., 2009, 11, 1346 RSC.
- R. King, J. Chem. Soc. Trans., 1921, 119, 2105 RSC.
- V. M. Mukkala, M. Helenius, I. Hemmilä, J. Kankare and H. Takalo, Helv. Chim. Acta, 1993, 76, 1361 CrossRef CAS.
- D. C. Duffy, J. C. McDonald, O. J. A. Schueller and G. M. Whitesides, Anal. Chem., 1998, 70, 4974 CrossRef CAS.
- S. C. Schafer and H.-A. Lehr, Pathobiology, 2007, 74, 259 Search PubMed.
- L. Aldwin and D. E. Nitecki, Anal. Biochem., 1987, 164, 494 CrossRef CAS.
- H. R. Nordlund, V. P. Hytonen, O. H. Laitinen, S. T. H. Uotila, E. A. Niskanen, J. Savolainen, E. Porkka and M. S. Kulomaa, FEBS Lett., 2003, 555, 449 CrossRef CAS.
- F. Song, J. Am. Soc. Mass Spectrom., 2007, 18, 1286 CrossRef CAS.
- M. Zehl and G. Allmaier, Anal. Chem., 2005, 77, 103 CrossRef CAS.
- M. Brinkley, Bioconjugate Chem., 1992, 3, 2 CrossRef CAS.
- A.-L. Gassner, C. Duhot, J.-C. G. Bünzli and A.-S. Chauvin, Inorg. Chem., 2008, 47, 7802 CrossRef CAS.
- M. Wilchek and E. A. Bayer, Anal. Biochem., 1988, 171, 1 CrossRef.
- M. D. Pierschbacher and E. Ruoslahti, Nature, 1984, 309, 30 CrossRef CAS.
- R. Molina, V. Barak, A. van Dalen, M. J. Duffy, R. Einarsson, M. Gion, H. Goike, R. Lamerz, M. Nap, G. Soletormos and P. Stieber, Tumor Biol., 2005, 26, 281 Search PubMed.
- H. Wang and J. Pevsner, Cell Tissue Res., 1999, 296, 511 CrossRef CAS.
- M. Albrecht, S. Schmid, S. Dehn, C. Wickleder, Z. Shuang, A. P. Bassett, Z. Pikramenou and R. Fröhlich, New J. Chem., 2007, 31, 1755 RSC.
- T. B. Jensen, R. Scopelliti and J.-C. G. Bünzli, Dalton Trans., 2008, 1027 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: formula of Eu-W8044 (Fig. S1), follow-up and optimization of the bioconjugation reaction (Fig. S2–S4, Table S1), crystal-field sublevels (Table S2), relative emission intensities (Table S3), immunoluminescence assays (Fig. S5, S6). See DOI: 10.1039/b922124g |
| ‡ Authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2010 |