Improving limits of detection for B-type natriuretic peptide using PC-IDMS: An application of the ALiPHAT strategy
Christopher M.
Shuford
a,
Daniel L.
Comins
a,
Jerry L.
Whitten
a,
John C.
Burnett, Jr.
b and
David C.
Muddiman
*a
aW.M. Keck FT-ICR Mass Spectrometry Laboratory, Department of Chemistry, North Carolina State University, Raleigh, North Carolina 27695, USA. E-mail: david_muddiman@ncsu.edu; Fax: +1 919-513-7993; Tel: +1 919-513-0084
bDivision for Cardiovascular Disease, Mayo Clinic College of Medicine, Rochester, Minnesota 55905, USA
First published on 19th November 2009
Abstract
Hydrophobic tagging of biomolecules has been reported by our group and others to increase their ionization efficiency during electrospray ionization and facilitate their detection by mass spectrometry. As such, hydrophobic tagging should provide a viable method for augmenting MS-based quantification of low abundance proteins by decreasing their detection limits. Herein we have evaluated two commercial alkylation reagents and several newly synthesized hydrophobic alkylation reagents for their utility in quantifying B-type Natriuretic Peptide, a low abundance cardiac biomarker, by protein cleavage isotope dilution mass spectrometry. For the cysteine containing tryptic peptide evaluated, a ∼3.5-fold decrease in the detection limit was observed for the best performing hydrophobic reagent, 2-iodo-N-octylacetamide, relative to the commonly used alkylation reagent, iodoacetamide. Additionally, we have evaluated the use of nonpolar surface areas as a metric for assessing the effectiveness of the alkylation reagents in improving ESI response.
Introduction
The first example of absolute protein quantification using mass spectrometry (MS) was reported by Barr et al. in 1996.1 In this study, the authors quantified apolipoprotein A-1 by subjecting the protein to enzymatic digestion and, then quantifying its proteolytic peptides using stable isotope-labeled (SIL) peptides as internal standards.1 Since this seminal report, protein cleavage isotope dilution mass spectrometry (PC-IDMS) has been adapted and refined in a number of publications.2–4 Stable isotope standards and capture by anti-peptide antibodies (SISCAPA) is a form of PC-IDMS developed by Anderson and coworkers where antibodies are applied to enrich targeted proteolytic peptides and their SIL counterparts prior to MS analysis.5 Beynon et al. have developed and applied recombinant peptide concatemers (QCAT) as SIL internal standards in order to multiplex PC-IDMS measurements.6 Recently, recombinant SIL proteins have been employed as internal standards in the protein standard absolute quantification (PSAQ) approach.7 Although PC-IDMS is now routinely used for absolute quantification of high and medium abundance proteins (ng/mL or greater),3,4 the concentrations of many biologically significant proteins are at or below the current detection threshold of MS-based technologies. As such, there is a need to improve the detection limits of PC-IDMS methods in order to better detect and quantify lower abundance proteins (pg/mL).The relationship between hydrophobicity and electrospray response are well established8–11 and hydrophobic tagging of biomolecules has been utilized to improve detection sensitivity during electrospray ionization (ESI) MS. Null et al. first demonstrated the utility of hydrophobic tagging to improve ESI response when they purposefully modified the 5′ terminus on a 20-mer oligonucleotide and a 53-bp PCR amplicon with an alkyl chain.11 This concept has since been adapted to modify peptides and proteins with hydrophobic moieties using various reagents that target guanidine groups12 or primary amines.13,14 Recently our group has developed a method, termed augmented limits of detection for peptides with hydrophobic alkyl tags (ALiPHAT), to increase the ionization efficiency of cysteine containing peptides and proteins.15,16 Albeit only applicable to cysteine containing peptides, the ALiPHAT method can be seamlessly integrated into traditional bottom-up proteomics experiments by simply using a more hydrophobic analogue of the common sulfhydryl reagent, iodoacetamide. More importantly, utilizing hydrophobic tagging to decrease detection limits should extend the utility of MS-based quantification to lower abundance proteins and facilitate targeted biomarker validation efforts.
Herein, the ALiPHAT strategy is applied to a PC-IDMS workflow to determine its efficacy for quantifying a low abundance cardiac biomarker, B-type Natriuretic Peptide (BNP). To date, very little molecular level information is known about the circulating forms and levels of endogenous BNP as they relate to the existence and severity of congestive heart failure. This lack of understanding is, in part, a result of the difficulties in detecting and characterizing BNP because it circulates in multiple forms and at extremely low concentrations.17 Our group provided the first molecularly specific evidence that the biologically active form of BNP (BNP-32) was decreased in heart failure using FT-ICR-MS.18 More recently, Schellenberger and co-workers have corroborated our results by using a novel immunoaffinity enrichment technique (termed MSIA) to quantify BNP-32 in CHF using MALDI-TOF-MS.19 Nonetheless, BNP-32 remains difficult to assay and detect by mass spectrometry. To this end, we have synthesized and evaluated an array of hydrophobic alkylation reagents as well as two commercially available reagents, iodoacetamide and N-ethylmalimide, in order to determine the optimal reagent for quantifying a tryptic peptide of BNP by PC-IDMS. Moreover, the implications of non-polar surface area (NPSA) on ESI response are evaluated for this cysteine containing peptide and compared to our previous findings, which have indicated that a species' ESI response can be diminished if it is made overly hydrophobic.
Experimental
Materials
All solvents used for these analyses were HPCL-grade and were purchased from Burdick and Jackson (Muskegon, MI). Formic acid, cysteine, iodoacetamide, N-ethylmaleimide, and Tris(2-carboxyethyl)phosphine (TCEP) were from Sigma-Aldrich (St. Louis, MO). Zwittergent 3–16 was purchased from Calbiochem (La Jolla, CA) and ammonium bicarbonate from Fisher Scientific (Fair Lawn, NJ). The hydrophobic alkylation reagents used herein were synthesized by the Comins laboratory at North Carolina State University using methods previously described.15Synthetic peptides for this study were purchased from the Mayo Proteomics Center (Rochester, MN) and were used as received. Both the natural (NAT) and stable-isotope labeled peptides had an amino acid sequence of ISSSSGLGCK, which corresponds to a tryptic peptide for BNP. The stable isotope-labeled version of this peptide incorporated a heavy leucine (13C615N) to provide a 7 Da mass shift relative to the natural version. Stock solutions of each peptide were made in water and their concentrations determined by UV-Vis Spectrophotometry and the Scopes method.20
Peptide modifications
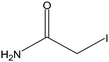
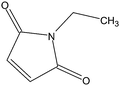
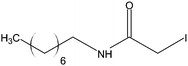
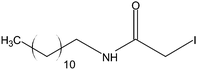
Stock solutions of the NAT and SIL peptides were combined to produce a series of peptide solutions with molar ratios (NAT:SIL) of 0:8, 1:8, 2:8, 4:8, 8:8, and 16:8. All peptide solutions were made in 200 mM ammonium bicarbonate buffer (pH 8.0) and had a SIL peptide concentration of 8 µM. Aliquots from each peptide solution were modified with one of eight alkylation reagents (Table 1), for a total of 48 reactions mixtures (6 peptide solutions × 8 alkylation reagents). For all reactions, a 50 µL aliquot of peptide solution was reduced with 40 µL of 100 µM TCEP (1 hr, 37 °C) and, subsequently alkylated with 96 µL of the respective alkylation reagent (1 hr, 37 °C). For the reagents IAM and NEM, 250 µM solutions were utilized for alkylation while 1000 µM solutions were used for the six hydrophobic reagents. Quenching of the alkylation reaction was performed by adding a 10-fold molar excess of cysteine to the reaction mixture (30 min, room temperature). Finally, the reaction mixture was diluted to a final volume of 500 µL to produce the modified-peptide solutions, which were stored at −20 °C until used. All reagents used for modification reactions were made fresh in 200 mM ammonium bicarbonate buffer (pH 8.0), with the exception of the hydrophobic tags which were made in a 50/50 mixture of buffer and methanol.| Alkylation Reagent | Natural Stable Isotope-Labeled | NPSA (Å)2 | |||
|---|---|---|---|---|---|
| Precursor m/z | Product m/z | ||||
| a The NPSA for these tags were incorrectly calculated in our previous report.22 It should be noted that these corrections are minor and do not affect the integrity or findings of the previously published work. | |||||
|
498.245 | → | 795.366 | ||
| IAM | 29 | ||||
| 501.754 | → | 802.383 | |||
|
532.258 | → | 863.392 | ||
| NEM | 82a | ||||
| 535.767 | → | 870.409 | |||
|
543.268 | → | 885.413 | ||
| OCAM | 192a | ||||
| 546.777 | → | 982.430 | |||
|
550.276 | → | 899.429 | ||
| DCAM | 268 | ||||
| 553.785 | → | 906.446 | |||
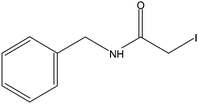
|
554.308 | → | 907.491 | ||
| PH1 | 115a | ||||
| 557.816 | → | 914.508 | |||
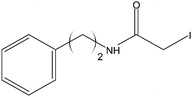
|
564.292 | → | 927.460 | ||
| PH2 | 134a | ||||
| 567.800 | → | 934.477 | |||
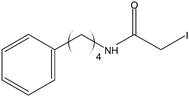
|
582.339 | → | 963.554 | ||
| PH4 | 172a | ||||
| 585.847 | → | 970.571 | |||
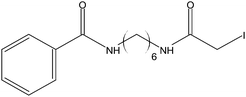
|
599.810 | → | 998.497 | ||
| BzAM | 210 | ||||
| 603.319 | → | 1005.514 | |||
NanoLC-SRM
Modified-peptide solutions were analyzed in two experimental sets. The first set consisted of the alkylation reagents iodoacetamide (IAM), N-ethylmaleimide (NEM), 2-iodo-N-octylacetamide (OCAM), and 2-iodo-N-dodecylacetamide (DCAM); and the second set consisted of 2-iodo-N-benzylacetamide (PH1), 2-iodo-N-(phenethyl)acetamide (PH2), 2-iodo-N-(4-phenylbutyl)acetamide, and N-(6-(2-iodoacetamido)hexyl)benzamide (BzAM). For each experimental set, the four modified peptide solutions having equivalent ratios of NAT and SIL peptides were combined to create equimolar mixtures of the various modified peptides. The resulting solutions were then diluted in 0.001% Zwittergent 3–16 (w/v) such that the concentration of any given modified SIL peptide was 800 pM. Lastly, solutions were stored at −20 °C until nanoLC-SRM analyses.All samples were thawed and analyzed in triplicate, in a randomized order with blank runs interdigitated. For all runs, 10 µL of each sample was injected by an HTS PAL Autosampler (LEAP Technologies, Carborro, NC) and loaded at 10 µL/min onto a custom built Magic C8 OPTI-PAK Trap Cartridge (Optimize Technologies, Oregon City, OR) using an LC-20AD high-flow pump (Shimadzu, Columbia, MD). After loading/desalting for 2.39 minutes in 100% mobile phase A (98/2/0.2, water/acetonitrile/formic acid), the 10-port switching valve (VICI, Houston, TX) was triggered to place the trap cartridge in-line with the analytical column for gradient elution. The analytical column consisted of a 75 µm i.d. PicoFrit capillary column (New Objective, Woburn, MA) with a 15 µm i.d. emitter tip packed in-house with 4 µm Jupiter Proteo C12 90 Å stationary phase (Phenomenex, Torrance, CA). Gradient elution was performed at 500 nL/min using a Chorus 220 nanoflow pump (CS Analytics, Zwingen, Switzerland). Initially, the gradient was held at 2% mobile phase B (98/2/0.2, water/acetonitrile/formic acid) and after 4 minutes, stepped immediately to 20% B. Over the next 14 minutes the gradient was ramped to 80% and then to 95% B in 1 minute. After maintaining at 95% B for 5 minutes the initial conditions were resumed and the column was allowed to re-equilibrate for 6 minutes prior to the next injection. Between runs, the syringe and injection port were rinsed thoroughly to minimize the effects of carryover.
Measurements were made on a TSQ Quantum Triple Quadrapole Mass Spectrometer (Thermo Scientific, San Jose, CA) externally calibrated according to the manufacturers protocol. The instrument was operated in SRM mode and was set to monitor the [M+2H+]2+ to y81+ transition for all modified peptides (see Table 1). Quadrupoles, Q1 and Q3, were operated with unit resolution and a 10 V skimmer offset was employed. Additionally, the ESI voltage, capillary offset and capillary temperature were set to 2000 V, 150 V and 250 °C, respectively.
Nano-LC-FT-ICR-MS
Each reaction mixture utilizing a 2:8 NAT:SIL molar ratio was diluted in 0.001% Zwittergent 3–16 to give a final SIL concentration of 25 nM. These samples were analyzed by online nano-LC-FT-ICR-MS utilizing an Eksigent 2D-nanoLC system (Dublin, CA) coupled to an LTQ-FT Ultra mass spectrometer from Thermo Scientific, Inc. (San Jose, CA). Online desalting/cleanup was achieved using the vented column configuration as previously described.21 With the exception of the valve configuration and loading flow-rate (3 µL/min) all chromatographic parameters were kept constant between the SRM and FT-ICR analysis. Tandem-MS spectra were acquired during the run using data-dependent MS/MS capabilities of the instrument.Nonpolar surface area
The NPSA for the BNP tryptic peptide (807 Å2) was calculated by summing the nonpolar surface areas for each amino acid residue as described by Cech and Enke.9 The NPSA for each alkylation reagent (see Table 1) was estimated as previously described using a combination of basic geometry, bond lengths, and van der Waals radii.22Results and discussion
In order to ensure the alkylation reactions were complete and localized to the peptide's lone cysteine residue, a reaction mixture for each reagent was analyzed by FT-ICR-MS. The resulting data were interrogated manually against in-silico alkylation products to identify the presence of the unalkylated peptide, the mono-alkylated peptide, and any overalkylated peptides. In all cases, the only peptides observed were the mono-alkylated species, signifying that all reactions resulted in complete modification. Additionally, analysis of tandem-MS spectra indicated the lone modification was isolated to the cysteine residue (data not shown). Thus, it was concluded that all modified peptides were present at the expected concentrations and the following observations were directly attributable to the modified peptides' inherent hydrophobicity, rather than varying reactivities of the different alkylation reagents.Representative chromatograms for each of the modified peptides are shown in Fig. 1 and, as expected, increased retention times correlated with an increase in NPSA of the modified peptides. One interesting exception to this was the BzAM-modified peptide, which eluted much earlier than anticipated based on its relatively large NPSA. The BzAM alkylation reagent is unique among the hydrophobic reagents in that it has an amide functional group in its alkyl tail. Although not taken into account by the NPSA calculation, the additional polar character afforded by the amide decreases the inherent hydrophobicity of the BzAM with respect to the other reagents and explains its unique chromatographic characteristic.
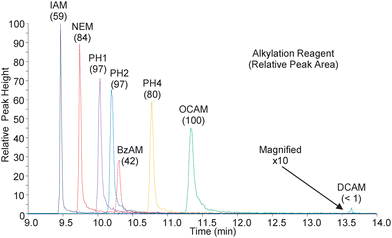 | ||
| Fig. 1 The above chromatograms were obtained during nanoLC-SRM analysis of the modified peptides. The transitions are for the natural version of the modified peptides with 8 femtomoles loaded on column. Shown in parentheses are the relative abundances as measured by the peak area. The DCAM modified peptide was magnified for visualization. | ||
Also noteworthy in Fig. 1 is the relationship between peak areas and peak widths. Despite the steep gradient employed, there is significant peak broadening observed across the narrow elution window and, consequently, the more retained species are less concentrated while eluting. Interestingly, increased peak areas were still observed for the majority of hydrophobic alkylation reagents, indicating the increased ionization efficiency of the more hydrophobic modified peptides was sufficient to overcome their poor chromatographic performance. This is important to note since the increase in abundances for the hydrophobically modified peptides is directly attributable to the improvement in ionization efficiency and not to improved enrichment/concentration of the peptide during the chromatographic separation. Indeed, the relative increase in signal for the more hydrophobic reagents may be more substantial with improved chromatography.
Fig. 2A shows the absolute abundance versus the amount of material loaded on column for each modified peptide. Except for the DCAM reagent, all the hydrophobic reagents and NEM yielded larger abundances than IAM across the concentration range. The fold improvement for each tag relative to IAM is shown in Fig. 2B. For the range plotted, the top performing alkylation reagents give between a 2- and 5-fold increase in abundance. The data points for 1 femtomole of material loaded on column are excluded in Fig. 2B because the signal for the IAM-modified peptide was below its calculated limits of detection (vida infra) and the resulting ratio was aberrantly large.
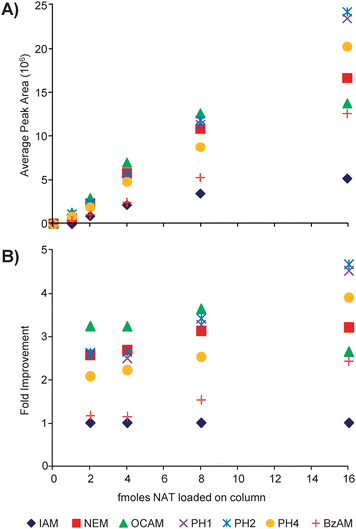 | ||
| Fig. 2 (A) Plotted are the average peak areas (n = 3) for the natural version of the modified peptides loaded on column in different amounts. (B) The average abundances of each modified peptide are normalized to that of the IAM-modified peptide (AX/AIAM) to determine the fold improvement. | ||
The PC-IDMS calibration curves in Fig. 3 were created for each modified peptide by plotting the peak area ratio (NAT/SIL) versus the molar ratio of material loaded on column (NAT/SIL) and performing linear least-squares regression analysis to generate a line of best fit. All alkylation reagents, with the exception of DCAM, yielded linear calibration curves with correlation coefficients ranging between 0.9845 and 0.9989. Additionally, all curves gave the expected slope of unity and intercepted the y-axis at the origin. The limits-of-detection (LOD) for each calibration curve were first calculated as the upper 95% confidence interval to the y-intercept as previously described.23 Using this value, the LOD in the x-domain (XLOD) was calculated using the respective least-squares regression equation. Finally, the product between XLOD and the amount of SIL internal standard loaded on column (8 femtomoles in all cases) was used to determine the LOD in terms of the amount of NAT peptide loaded on column. These resulting values are plotted in Fig. 4 along with the fold improvement relative to the IAM-modified peptide. For the alkylating reagents evaluated, all gave a minimum 2-fold improvement in detection limits relative to IAM. The OCAM-modified peptide performed best giving an LOD of 360 attomoles loaded on column (or 125 pg/mL), which was ∼3.5 fold better than the IAM-modified peptide at 1260 attomoles loaded onto the column (or 436 pg/mL). In the context of physiological BNP, the biologically active form of BNP (i.e. BNP-32) is estimated to circulate at 1–111 pg/mL in stage I; 111–235 pg/mL in stage II; 235–461 pg/mL in stage III; and greater than 461 pg/mL in stage IV NYHA congestive heart failure patients.24 Although the reported detection limits do not reflect what is necessarily obtainable in a real sample (e.g., in plasma), they suggest that by simply switching from a standard alkylation reagent to an appropriate hydrophobic reagent the utility of PC-IDMS measurements for BNP-32 can be extended from stage III to stage II patients. Moreover, this method should be applicable for a number of cysteine containing proteins, so long as their proteolytic peptides meet the necessary criteria for PC-IDMS.
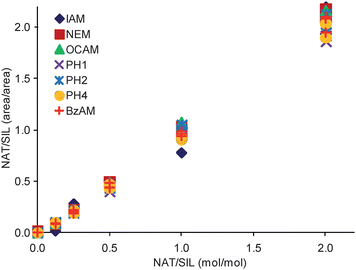 | ||
| Fig. 3 Calibration curves for each modified peptide are shown. The slopes for the displayed curves ranged between 0.9584 and 1.0787, while the intercepts were between −0.0523 to −0.0202. | ||
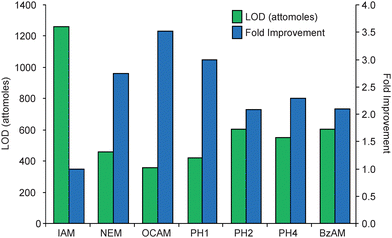 | ||
| Fig. 4 The LOD for each modified peptide is displayed in the bar graph as attomoles of modified peptide loaded on column. To determine the fold improvements, the LOD for each species was normalized to the IAM-modified peptide (LODIAM/LODX). In terms of the total amount of material loaded on column, the LODs for IAM, NEM, OCAM, PH1, PH2, PH4, and BzAM modified peptides were 1260, 457, 357, 420, 603, 549, and 601 attomoles, respectively. Since DCAM did not provide a linear calibration curve, its detection limit could not be reliably calculated. | ||
Previous reports have indicated that modification of peptides with hydrophobic alkylation reagents can increase the hydrophobicity of the peptide to a point where it is detrimental for ionization.16,22 To evaluate this phenomenon, our group has developed a metric that relates the NPSA of a modified peptide with the observed abundance.22Fig. 5 demonstrates this relationship for the BNP tryptic peptide by plotting the NPSA of each modified peptide versus the average observed abundance for 8 femtomoles loaded on column. Consistent with previous findings, there appears to be an optimal NPSA range in which maximum ESI response is achieved (roughly 900 to 1000 Å2). Moreover, in our recent report22 a maximum ESI response for Laminin nanopeptide and the N-terminal tryptic peptide of prostate specific antigen was achieved in a nearly identical NPSA range as what was observed in this independent study. Although not conclusive because of the small sample size (3 peptides),22 the combined data indicates there may be an absolute NPSA range in which all peptides will achieve their greatest ESI response. This could be particularly useful as hydrophobic alkylation reagents could be designed with the appropriate characteristics to place any cysteine containing peptide within the optimal NPSA range. Likewise, in cases where peptides are overly hydrophobic, such as in membrane proteins, it should be feasible to modify them with a hydrophilic reagent to decrease their NPSA and optimize their ESI response.
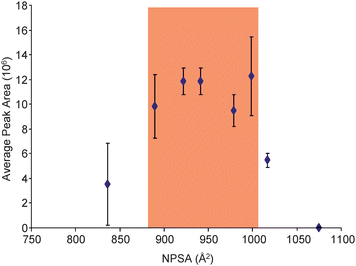 | ||
| Fig. 5 Plotted are the average peak areas (n = 18) for the SIL version of the modified peptides with 8 femtomoles loaded on column versus their respective NPSAs. The NPSA for each modified peptide was estimated by summing the NPSA for the tryptic peptide and the respective reagent. Highlighted is the NPSA range that appears to provide the optimal ESI response for the BNP tryptic peptide. | ||
Conclusions
PC-IDMS calibration curves were developed for a cysteine containing tryptic peptide of the cardiac biomarker, BNP. Several hydrophobic alkylation reagents developed for the ALiPHAT method, and two commercially available reagents were evaluated for their utility in generating these curves. With the exception of the DCAM reagent, all tags were observed to react completely and specifically with the target peptide and also to generate viable calibration curves. The more hydrophobic reagents improved the LOD for the measurement of the BNP tryptic peptide, demonstrating the ability of the ALiPHAT method to expand the utility of PC-IDMS measurements for low abundance proteins. Additionally, the NPSA metric applied to this peptide was consistent with data from our previous report. The findings imply there is an absolute NPSA range in which all peptides have a maximum ESI response and, consequently, a minimum LOD.Acknowledgements
The authors are appreciative of the financial support from the National Institutes of Health (Grant 5RO1HL036634) and the NIH/NCSU Molecular Biotechnology Training Program (Grant 5T32GM00-8776-08), which supports CMS.References
- J. R. Barr, V. L. Maggio, D. G. Patterson, G. R. Cooper, L. O. Henderson, W. E. Turner, S. J. Smith, W. H. Hannon, L. L. Needham and E. J. Sampson, Clin. Chem., 1996, 42, 1676–1682 CAS.
- S. A. Gerber, J. Rush, O. Stemman, M. W. Kirschner and S. P. Gygi, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6940–6945 CrossRef CAS.
- K. Kito and T. Ito, Curr. Genomics, 2008, 9, 263–274 CrossRef CAS.
- S. Pan, R. Aebersold, R. Chen, J. Rush, D. R. Goodlett, M. W. McIntosh, J. Zhang and T. A. Brentnall, J. Proteome Res., 2009, 8, 787–797 CrossRef CAS.
- N. L. Anderson, N. G. Anderson, L. R. Haines, D. B. Hardie, R. W. Olafson and T. W. Pearson, J. Proteome Res., 2004, 3, 235–244 CrossRef CAS.
- R. J. Beynon, M. K. Doherty, J. M. Pratt and S. J. Gaskell, Nat. Methods, 2005, 2, 587–589 CrossRef CAS.
- V. Brun, A. Dupuis, A. Adrait, M. Marcellin, D. Thomas, M. Court, F. Vandenesch and J. Garin, Mol. Cell. Proteomics, 2007, 6, 2139–2149 CrossRef CAS.
- J. B. Fenn, J. Am. Soc. Mass Spectrom., 1993, 4, 524–535 CrossRef CAS.
- N. B. Cech and C. G. Enke, Anal. Chem., 2000, 72, 2717–2723 CrossRef CAS.
- N. B. Cech and C. G. Enke, Anal. Chem., 2001, 73, 4632–4639 CrossRef CAS.
- A. P. Null, A. I. Nepomuceno and D. C. Muddiman, Anal. Chem., 2003, 75, 1331–1339 CrossRef CAS.
- A. Foettinger, A. Leitner and W. Lindner, J. Mass Spectrom., 2006, 41, 623–632 CrossRef CAS.
- H. Mirzaei and F. Regnier, Anal. Chem., 2006, 78, 4175–4183 CrossRef CAS.
- R. Ullmer, A. Plematl and A. Rizzi, Rapid Commun. Mass Spectrom., 2006, 20, 1469–1479 CrossRef CAS.
- J. L. Frahm, I. D. Bori, D. L. Comins, A. M. Hawkridge and D. C. Muddiman, Anal. Chem., 2007, 79, 3989–3995 CrossRef CAS.
- D. K. Williams, C. W. Meadows, I. D. Bori, A. M. Hawkridge, D. L. Comins and D. C. Muddiman, J. Am. Chem. Soc., 2008, 130, 2122 CrossRef CAS.
- F. S. Apple, A. H. B. Wu, A. S. Jaffe, M. Panteghini and R. H. Christenson, Clin. Biochem., 2008, 41, 222–226 CrossRef CAS.
- A. M. Hawkridge, D. M. Heublein, H. R. Bergen, A. Cataliotti, J. C. Burnett and D. C. Muddiman, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17442–17447 CrossRef CAS.
- E. E. Niederkofler, U. A. Kiernan, J. O'Rear, S. Menon, S. Saghir, A. A. Protter, R. W. Nelson and U. Schellenberger, Circulation Heart Failure, 2008, 1, 258–264 Search PubMed.
- R. K. Scopes, Anal. Biochem., 1974, 59, 277–282 CrossRef CAS.
- G. L. Andrews, C. M. Shuford, J. C. Burnett, A. M. Hawkridge and D. C. Muddiman, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2009, 877, 948–954 CrossRef CAS.
- D. K. Williams, D. L. Comins, J. L. Whitten and D. C. Muddiman, J. Am. Soc. Mass Spectrom., 2009, 20, 2006–2012 CrossRef CAS.
- I. Lavagnini and F. Magno, Mass Spectrom. Rev., 2007, 26, 1–18 CrossRef CAS.
- A. S. Maisel, P. Krishnaswamy, R. M. Nowak, J. McCord, J. E. Hollander, P. Duc, T. Omland, A. B. Storrow, W. T. Abraham, A. H. B. Wu, P. Clopton, P. G. Steg, A. Westheim, C. W. Knudsen, A. Perez, R. Kazanegra, H. C. Herrmann and P. A. McCullough, N. Engl. J. Med., 2002, 347, 161–167 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |