Cadmium binding in mixtures of phytochelatins and their fragments: A voltammetric study assisted by multivariate curve resolution and mass spectrometry
Rui
Gusmão
,
Cristina
Ariño
,
José Manuel
Díaz-Cruz
and
Miquel
Esteban
*
Departament de Química Analítica, Facultat de Química, Universitat de Barcelona, Martí i Franquès 1 - 11, E - 08028, Barcelona, Spain. E-mail: miquelestebanc@ub.edu; Fax: +34 93 402 12 33; Tel: +34 93 403 91 17
First published on 13th November 2009
Abstract
Phytochelatins (PCn, (γ-Glu-Cys)nGly) are cysteine-rich peptides synthesized by plants which are involved in metal bioregulation and phytoremediation. Multivariate Curve Resolution by Alternating Least Squares (MCR-ALS) is applied to voltammetric data obtained from the analysis of the competitive binding of Cys or Cys-Gly with PC2 or PC3 by Cd2+. The displacements between ligands, the chain length dependence on the competitive binding to PCn and the possible existence of mixed ligand metal-complexes are investigated. The shape analysis of the resulting pure voltammograms and concentration profiles of the components resolved by MCR-ALS suggests that ligands containing more thiol groups are able to displace the shorter chain ligands from their metal complexes, whereas the opposite does not happen. Electrochemical results are compared with ESI-MS measurements.
1. Introduction
Nonessential heavy metal ions (e.g. Cd2+) are highly reactive and consequently can be toxic to living cells,1 leading to deregulation of cell cycle patterns. In the majority of cases Cd induces damage involving free radical generation that alters mitochondrial integrity and triggers apoptosis (programmed cell death).2,3 Thus plants, like other living organisms, have developed a variety of mechanisms that control and respond to the uptake and accumulation of both essential and nonessential heavy metals. These mechanisms include the chelation and sequestration of heavy metals by particular ligands. The two best characterized heavy metal-binding ligands in living cells are the phytochelatins (PCn) and metallothioneins (MT).3PCn consists of three aminoacids, viz. cysteine, glycine and glutamic acid, arranged in a (γ-GluCys)n-Gly conformation, where n is generally in the range of 2 to 6.4 PCn, as well as glutathione (γ-Glu-Cys-Gly, GSH), its precursor in the biosynthetic pathway, and other Cys-rich fragments (e.g. Cys-Gly), are capable of binding metal ions, mainly via thiolate coordination, and the resulting complexes may be stored in the vacuole or exported to the shoot via the xylem.5,6 This is one of the possible mechanisms through which plants can clean up and restore damaged ecosystems, the basis of phytoremediation approaches3,7,8 – a subject that has gained great interest with the possibility of using transgenic plants.9,10
In Cd-PCn systems, Cd can bind one, two, three or, at maximum capacity, four sulfurs from either single or multiple PCn molecules.11 However, the sequence of formation and the final stoichiometry of the different PCn–metal complexes involved remain largely unknown. For this reason, it is of great interest to study the complexation mechanisms in order to understand and optimise processes of phytoremediation. Several analytical approaches have been developed: atomic absorption spectrometry (AAS),12 atomic fluorescence spectrometry (AFS),13 and inductively coupled plasma-mass spectrometry (ICP-MS)14 are frequently used for the screening and quantification of Cd-containing fractions. Electrospray ionization-mass spectrometry (ESI-MS),15–17 circular dichroism spectroscopy (CD)18 and nuclear magnetic resonance (NMR)19 have also been used for the structural identification of the heavy metal complexes of Cys-rich peptides.
Polarographic and voltammetric techniques, especially in pulse mode, have proved to be a powerful tool to obtain information on physico-chemical properties of heavy metal complexes with PCn or GSH,20–22 since they provide different signals for the free peptide, the free metal ion and the bound metal in different chemical environments, and also allow for direct detection of thiols.23–26 The detailed mechanism of anodic reactions of thiol groups on mercury electrodes has been described by the use of different voltammetric techniques.24–26 The reactions consist of electro-oxidation of mercury in two separate steps to mercurous and mercuric thiolates which are both strongly adsorbed at the electrode. As a result, several anodic signals are usually obtained in the voltammetric study of metal-thiolate systems. This can cause additional interpretation difficulties when they overlap with characteristic signals of the considered species, although in some cases they allow us to follow the evolution of free thiolate groups. Discrimination between thiols and their metal complexes, therefore, is far from facile with minor shifts in potential effectively preventing voltammetric resolution of the peaks.
A valuable improvement of electrochemical methods is achieved when these are assisted by powerful chemometrical tools, such as Multivariate Curve Resolution with Alternating Least Squares (MCR-ALS).27–30 The use of chemometrics in electroanalytical chemistry is not as popular as in spectroscopy,31 although recently the application of these methods for mathematical resolution of overlapping signals, calibration and model identification has increased.29,30 The implementation of new restrictions such as peak shape, and more recently, signal shift correction,32 adapted to the special character of electrochemical signals, has allowed the resolution of quite intricate metal complex systems.
A great deal of knowledge has been obtained by these means for systems involving one metal ion plus a unique ligand,33–39 among which Cys-rich peptides33,34,39 and PCn35–39 are included. Although differential pulse polarography (DPP)/MCR-ALS may not always allow us to propose structures for the complexes, the stoichiometries deduced from the MCR-ALS concentration profiles provide essential information on the composition of complexes that can be complemented with non-electrochemical techniques such as ESI-MS37,39 or isothermal titration calorimetry (ITC).37 Another strategy to solve some uncertainties is the simultaneous MCR-ALS analysis of a data matrix containing the experiments carried out under the same conditions by different techniques such as, for instance DPP and CD.38 The complexity of the voltammograms obtained in these titrations prevents their direct interpretation using traditional electrochemical methods. But when MCR-ALS is applied, voltammograms can be interpreted rather satisfactorily, which makes them useful for the metal speciation of environmentally relevant natural ligands.
In a previous paper,39 the competition between two different GSH fragments (Cys and Cys-Gly) for heavy metal binding was investigated, using the combination of DPP and MCR-ALS. In that study two general conclusions were reached: i) no evidence of formation of mixed ligand complexes was found, and ii) a fast exchange equilibrium of the metal among the different ligands was observed, since the short time between consecutive additions proved to be sufficient to displace the metal in order to form the complexes that a priori appeared to be more stable.
In the present work, the competitive binding of Cd2+ towards systems consisting of PCn and one of its fragments is studied by DPP/MCR-ALS: i) Cys-Gly and PC2; ii) Cys and PC3. Fig. 1 summarizes the structure of all these peptides. In the previous paper, the study of the competitive binding was made with two GSH fragments with one thiol group. Here, the competitive binding of Cd is made between Cys-rich peptides (of different chain length) with a single thiol group and a PCn (n = 2,3), with an increasing number of thiol groups. In this study the displacements between ligands and the dependence of chain length on the transfer of the metal ion to PCn are analyzed. Additionally, the possibility of formation of mixed ligand metal-complexes, due to the typical tetrahedral coordination of Cd2+ that can be achieved with thiol groups of different peptides, is investigated.
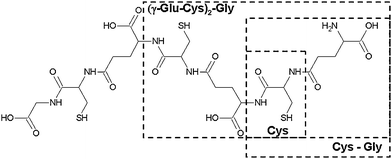 | ||
| Fig. 1 PC3 structure with the studied fragments highlighted (Cys, Cys-Gly and PC2). | ||
2. Experimental
2.1 Experimental and instrumentation
L-Cysteine (free base) and Cys-Gly (fluoroacetate salt) were provided by Sigma with a minimum purity of 98.0 and 85.0% respectively. The phytochelatins (γ-Glu–Cys)n-Gly (n = 2,3), as trifluoroacetate salts, were provided by DiverDrugs S.L. (Barcelona, Spain) with a purity ranging from 96.9% to 99.0%. Stock solutions (1 mL) of 1 × 10−3 mol L−1 thiol peptides were prepared daily. To ensure homogeneity, peptide solutions were mixed at 1400 rpm for 1 min using Eppendorf Mixmate.All other reagents were analytical-reagent grade from Merck. Cd2+ stock solution was prepared from Cd(NO3)2·4H2O and standardized complexometrically. TRIS-HNO3 ([tris(hydroximethyl)aminomethane]-nitric acid) was used as pH 7.5 buffer, and KNO3 was used as a supporting electrolyte at 0.1 mol L−1 to set ionic strength.
Polarographic measurements were performed in a Metrohm-757 VA Computrace attached to a personal computer with data acquisition software also from Metrohm. Working, reference and auxiliary electrodes were a static mercury drop electrode (SMDE) with a drop area of 0.9 mm2, an Ag/AgCl, KCl (3 mol L−1), and a glassy carbon respectively, all of them from Metrohm. Double distilled Hg was used.
Instrumental parameters for DPP experiments were a drop time of 1 s, scan rate of 0.005 V s−1, pulse time of 0.05 s, and pulse amplitude of 50 mV.
Positive-mode ESI-MS experiments for mixtures of Cd2+ with Cys-Gly and PC2, or Cys and PC3, were performed using a Q-TOF Agilent 1100 instrument.
pH measurements were made with an Orion SA 720 pH-meter. All the experiments were carried out at 25 °C and purified nitrogen was used for solution deaeration.
2.2 Procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 acetonitrile (HPLC grade) −5 × 10−3 mol L−1 ammonium acetate mixture was adjusted at pH 7.5 and used as a mobile phase.
:9 acetonitrile (HPLC grade) −5 × 10−3 mol L−1 ammonium acetate mixture was adjusted at pH 7.5 and used as a mobile phase.
3. Data treatment
Polarograms were smoothed, baseline corrected and converted into data matrices using home-made programs implemented in MATLAB.40 MCR-ALS analysis of data was carried out through several programs also implemented in MATLAB (some of them available at http://www.ub.edu/mcr/ndownload.htm), following the general methodology described in previous papers.33–38 In this work, the constraints used were non-negativity and signal-shape.Briefly, in the MCR-ALS analysis the matrices of currents I (nR, nE) are composed from the set of experimental voltammograms, which contain as many nR rows as the number of voltammograms recorded at each metal-to-ligand ratio and as many nE columns as potentials scanned in every run. After the smoothing and background correction, the matrix I is mathematically decomposed into the product of two factor matrices
| I = CVT + X | (1) |
At this point it is crucial to indicate the meaning of the concept of component (related to MCR-ALS) in the present context. For spectroscopic data, a component is associated to pure chemical species in solution, but for electrochemical data a component must be defined as a single electrochemical process giving a signal,27 including not only redox processes but also other possible phenomena, such as electrode adsorption of species or capacitive currents due to the charging of the electrical double layer at the electrode surface. In any case, most situations, a single electrochemical process is produced by a single species, but in some others a single species can produce two or more simple electrochemical processes, in which case an accurate analysis must be done to correlate components (or processes) with chemical species.
4. Results and discussion
4.1 Cd/Cys-Gly/PC2 system
The competitive complexation process of Cys-Gly and PC2 towards Cd2+ was studied by means of DPP titrations of different combinations of the three species. Fig. 2a–c show the evolution of the DPP curves when one of the ligands is added to solutions containing partially free and/or bound (to the other ligand) Cd2+. From these plots different groups of signals can be distinguished: (i) signals at more negative potentials than −0.65 V that are due to the reduction of the different complexes formed; (ii) signals at more positive potentials than −0.65 V related to the anodic oxidation of Hg in the presence of ligands and; (iii) a well defined peak at −0.63 V that corresponds to the free Cd2+ reduction.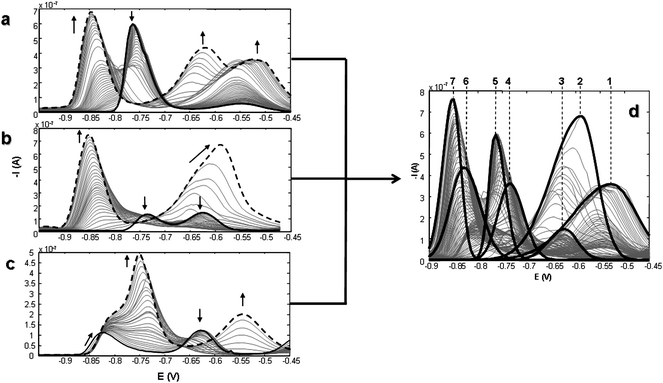 | ||
Fig. 2 Evolution of DPP curves along PC2 or Cys-Gly additions to Cd/Cys-Gly or Cd/PC2 solutions respectively, in 0.1 mol L−1 TRIS-HNO3 buffer (pH 7.5): a) PC2 is added to a 1:2 Cd:Cys-Gly solution; b) PC2 is added to a 2:1 Cd:Cys-Gly solution; c) Cys-Gly is added to a 3:1 Cd:PC2 solution. Arrows indicate the tendency of the signal (— initial DPP curve; ![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) final DPP curve). Unitary concentration: 1 × 10−5 mol L−1. d) Initial estimation of components. Numbers in figure indicate the components involved in the reduction process: Cys-Gly (1), PC2 (2), Cd2+ (3), Cd/Cys-Gly complexes (4 and 5) and Cd/PC2 complexes (6 and 7). final DPP curve). Unitary concentration: 1 × 10−5 mol L−1. d) Initial estimation of components. Numbers in figure indicate the components involved in the reduction process: Cys-Gly (1), PC2 (2), Cd2+ (3), Cd/Cys-Gly complexes (4 and 5) and Cd/PC2 complexes (6 and 7). | ||
When PC2 is added to 1:2 Cd2+:Cys-Gly solution, i.e. all metal ions initially complexed by the ligand (Fig. 2a), a decrease of the Cd/Cys-Gly signals (Ep = −0.76 V) combined with the appearance of a shoulder at more negative potentials than −0.80 V is observed. With further additions of PC2, this shoulder develops into a very well defined signal (Ep = −0.85 V) at the end of the titration. Simultaneously an increase of the anodic oxidation of Hg signal at less negative potentials takes place (ca. −0.55 V). This behaviour seems to underline the fact that PC2 displaces Cd2+ from the Cd/Cys-Gly complexes generating new and more stable Cd/PC2 complexes.
When PC2 is added to a 2:1 Cd2+:Cys-Gly solution (Fig. 2b), free Cd2+ and Cd/Cys-Gly complexes are initially present giving two overlapping peaks. Successive additions of PC2 produce a decrease of these signals in different proportions and an increase of the peak corresponding to Cd/PC2 complexes (at −0.85 V) together with the characteristic wide signals of the ligands (at −0.65 < E < −0.5 V). In this case, a remarkable movement in the ligand peak (PC2) is observed, which could cause additional difficulties in the chemometrical treatment.
An analogous situation, in which Cys-Gly is added to a 3:1 Cd:PC2 mixture (Fig. 2c) was also studied; such a situation is characterized by the presence of two signals at the beginning of the titration: one is related with Cd/PC2 complexes (Ep = −0.83 V), another with the free Cd2+ reduction (Ep = −0.63 V). Additions of Cys-Gly produce the depletion of the free Cd2+ and the formation of the successive complexes with Cys-Gly, which coexist with the Cd-PC2 previously present in solution. At the end of the titration, an intense and asymmetric signal related to the presence of the new Cd/Cys-Gly complex and the previously existing Cd/PC2 complex is observed. Simultaneously, the characteristic signals related with the anodic oxidation of Hg in the presence of free Cys-Gly (Ep = −0.55 V) is observed. In this experiment, the characteristic signal of free PC2 is absent, which is an indicator that the Cd/PC2 complex remains throughout Cys-Gly titration.
In addition, Cys-Gly is added to a 1:2 Cd2+:PC2 solution. Initially, all Cd2+ ions are chelated by PC2, which yields a well defined signal at Ep = −0.84 V; this signal does not change with the successive additions of Cys-Gly, as would be expected, due to the greater stability of Cd-PC2 complexes (results not shown). The increase of the signal at a less negative potential (ca. −0.55 V) is the only change observed in these voltammograms. This signal is related to the anodic oxidation of Hg in the presence of free Cys-Gly.
In order to analyze this behaviour (Fig. 2a–c) by MCR-ALS, voltammetric data were organized in a combined column-wise matrix, i.e., sharing the same unit voltammograms but different concentration profile matrices. An initial estimation of components is necessary for the iterative ALS optimization. This estimation has been built with the peakmaker subroutine, considering the facts described above and the information from previous papers on the complexation processes of Cd/Cys-Gly34 and Cd/PC2.35 Seven components are necessary for the proper resolution of data as Fig. 2d shows (components have been ordered considering their position on the potential axis). Some, or all, of them are present in the different titration experiments. At this point, it is important to reiterate that in voltammetric measurements, components are related to electrodic processes that do not necessarily correspond to chemical species as in the case of spectroscopic measurements.27
The number of components of each individual Cd/L (L = Cys-Gly or PC2) matrix should be enough to explain the evolution of signals in the extended column-wise matrix. Therefore, components 1 and 2 correspond to the signal of free ligands, Cys-Gly (Ep = −0.52 V) and PC2 (Ep = −0.60 V) respectively, component 3 to the reduction of free Cd2+ (Ep = −0.63 V), while the reduction of Cd2+ bound to Cys-Gly is shown by the presence of components 4 (Ep = −0.74 V) and 5 (Ep = −0.76 V). From previous studies34 these components could be associated with ML and ML2 complexes respectively. Reduction of Cd2+ bound to PC2 yields components 6 and 7, at −0.83 and −0.86 V respectively.
Constraints of non-negativity (for both concentrations and signals), signal shape (for the voltammograms of all species) and selectivity (depending on the submatrix) have been applied to the MCR-ALS iterative process that yields unitary signals that are almost identical to and centred at the same potential as those used as initial estimation (Fig. 2d) and the concentration profiles shown in Fig. 3a–c, with a lack of fit (lof) of 15.04%. It is important to note the great power of the MCR-ALS in the resolution of combined matrices, that, considering such a high number of components, yields acceptable lof values.
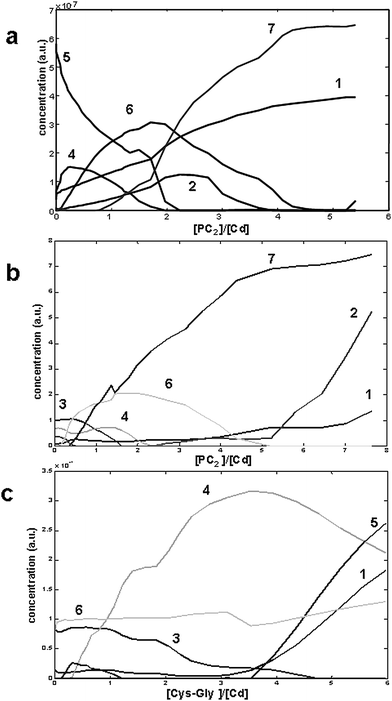 | ||
| Fig. 3 Respective concentration profiles obtained in the MCR-ALS decomposition of the combined column-wise matrix of data shown in Fig. 2a–c. Applied constrains were non negativity and signal shape. The concentration profiles are plotted as function of the added ligand-to-ligand (a) or ligand-to-metal concentration ratios (b and c). Components are: Cys-Gly (1), PC2 (2), Cd2+ (3), Cd/Cys-Gly complexes (4 and 5) and Cd/PC2 complexes (6 and 7). | ||
Concentration profiles (Fig. 3a) of the sub-matrix in Fig. 2a show that components 4 and 5 (Cd/Cys-Gly complexes) suffer a progressive decrease from the first addition of PC2. Parallel to this, component 6 rises and reaches a maximum at titrating ligand-to-Cd2+ ratios quite higher than what would be the expected ratio in a titration in the absence of Cys-Gly. This higher ratio could be due to the fact that the exchange equilibrium between ligands is not totally displaced, which is evidenced by the smooth decrease of components 4 and 5. Total displacement requires an additional PC2 excess to reach the same 1:1 stoichiometry for the individual Cd2+-PC2 system. This behaviour suggests a rapid transfer of Cd2+ ions between peptides and, seemingly, the absence of mixed complexes. From ratios higher than 1 to the end of the titration, component 7 increases. This is due to the formation of a more stable complex with a signal similar to that of the 1:2 Cd/PC2 complex observed in the absence of Cys-Gly. Component 1 increases throughout the titration which is consistent with the idea of PC2 capturing the Cd2+ ions complexed with Cys-Gly. MCR-ALS results fails to explain free PC2 evolution (component 2) due to the ambiguity with the signal of Cys-Gly.
For the sub-matrix in Fig. 2b, the best MCR-ALS results were obtained by assuming the absence of component 5 (ML2, L = Cys-Gly). The concentration profile (Fig. 3b) supports the idea that PC2 binds first to the free Cd2+ (component 3), subsequently displaces and binds Cd2+ from Cd/Cys-Gly complex (component 4), yields Cd/PC2 complexes (components 6 and 7) at more negative potentials and finally remains in excess as component 2. Component 6 achieves a maximum at a PC2-to-Cd ratio very similar to that of the previous experiment, and component 7 starts to increase at ratios lower than in the previous case, probably favoured by the initial presence of free Cd2+.
Concentration profiles obtained from the MCR-ALS analysis of the submatrix in Fig. 2c (Fig. 3c), by assuming the absence of components 2 and 7, give evidence that Cys-Gly binds only the free Cd2+ ions (component 3), giving successively the ML and ML2 complexes (components 4 and 5 respectively), at less negative potentials than the initially present Cd-PC2 complex (component 6). The concentration profile of this component does not change throughout the titration although it is somewhat affected by baseline problems. Once again, the added Cys-Gly does not have the capability of capturing the Cd2+ ions chelated by PC2. Component 6 is stable throughout the titration.
To complete the competition study of Cys-Gly and PC2 towards Cd complexation, successive aliquots of Cd2+ solution are added to an equimolar mixture of Cys-Gly and PC2 (Fig. 4a). This type of titration (a ligand mixture with a metal ion that is initially absent in solution) is of great interest because it allows us a better prediction of the behaviour of a natural system when it is exposed to metal ion stress or contamination. Comparison between each individual ligand titration with Cd2+ (Fig. 4b) shows that signal evolution is very similar, reinforcing the concept of a greater stability of Cd/PC2, complexes in comparison with those of Cd and Cys-Gly, and the absence of mixed ligand metal complexes.
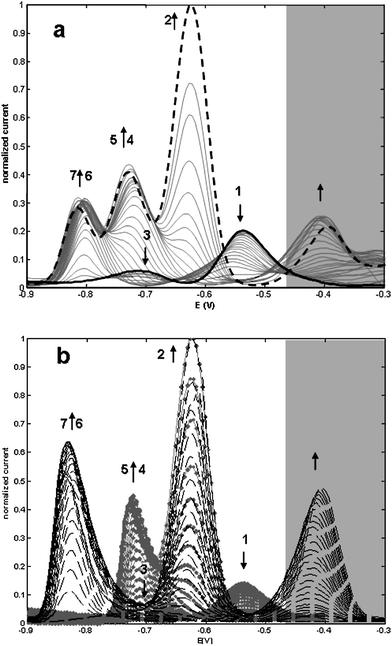 | ||
Fig. 4 Evolution of DPP curves along Cd2+ additions to a 1:1 Cys-Gly:PC2 solution (a) (— initial DPP curve; final DPP curve). Evolution of DPP curves along Cd2+ additions of independent titrations (b) of 1 × 10−5 mol L−1 Cys-Gly (![[dash dot, graph caption]](https://www.rsc.org/images/entities/char_e090.gif) ) or 1 × 10−5 mol L−1 PC2 (). All experiments at pH 7.5 in 0.1 mol L−1 TRIS-HNO3 buffer in the presence of KNO3 0.1 mol L−1. Arrows indicate the evolution of the signals. ) or 1 × 10−5 mol L−1 PC2 (). All experiments at pH 7.5 in 0.1 mol L−1 TRIS-HNO3 buffer in the presence of KNO3 0.1 mol L−1. Arrows indicate the evolution of the signals. | ||
Prior to the addition of Cd2+, two signals related with the anodic oxidation of Hg in the presence of Cys-Gly (component 1, Ep = −0.50 V) and PC2 (component 3, Ep = −0.70 V) are observed (continuous bold line in Fig. 4a). Successive additions of Cd2+ to the 1:1 Cys-Gly:PC2 mixture clearly produce three wide signals: i) one that corresponds to the Cd2+ reduction peak (component 2, Ep = −0.63 V); ii) one to the formation of Cd/Cys-Gly complexes (components 4 and 5, at ca. −0.70V); and iii) the last one, at potentials around −0.8 V, that corresponds to PC2 complexes (components 6 and 7). Close to −0.40 V, a signal that can be assigned to the anodic process of Hg in the presence of Cd/PC2 complexes is observable (see Fig. 4b, dashed lines). This signal is excluded from MCR-ALS treatment, because non-relevant information can be obtained from it.
Following the same criteria, and taking into account the initial estimation of components used to explain individual titrations, MCR-ALS produced a reliable resolution of the data matrix in Fig. 4a (lof = 9,84%) when the constraints of non-negativity, for both concentrations and signals, and signal shape, for voltammograms of all components, are applied. Additionally, selectivity was applied to component 3, forcing it to a zero value once it reached null value. Unitary voltammograms and concentration profiles are represented in Fig. 5. In a first analysis, it is observable that the represented profile does not differ essentially from the concentration profiles of a PC2 titration with Cd2+35. Concentration profiles (Fig. 5b) show that PC2 (component 3) reaches a minimum faster than Cys-Gly (component 1). However this last component does not reach a null value, probably due to the overlapping of these signals with those related to the anodic oxidation of Hg in the presence of Cd/PC2 complexes.
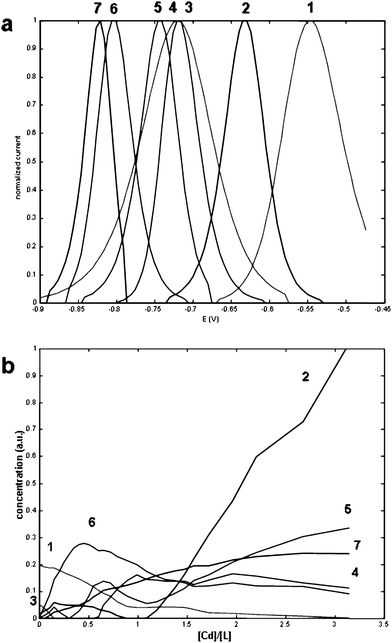 | ||
| Fig. 5 Unitary voltammograms (a) and concentration profiles (b) obtained in the MCR-ALS decomposition of the data matrix shown in Fig. 4a. Applied constrains were non negativity, signal shape and selectivity for ligands at the beginning of the titration. The concentration profiles are plotted as function of the added Cd2+-to-ligand concentration ratios (L = Cys-Gly or PC2). Components are: Cys-Gly (1), Cd2+ (2), PC2 (3), Cd/Cys-Gly complexes (4 and 5) and Cd/PC2 complexes (6 and 7). | ||
These results also show that the presence of Cys-Gly does not affect the formation of Cd/PC2 complexes. Only when component 6 (ML2, L = PC2) reaches its maximum (at ratio around 0.5) do components 4 and 5 (related to ML2 and M2L2, L = Cys-Gly) start to appear. Furthermore, only when component 7 reaches a plateau (M2L2, L = PC2) for ratios greater than 1.5, is a consistent increase in the signal of component 5 (M2L2, L = Cys-Gly) observable. This result expresses the greater capacity of PC2 to bind Cd2+, in comparison with a smaller ligand with a single thiol group.
Electrochemical results were compared with those obtained by ESI-MS for a 3:1:1 Cd/Cys-Gly/PC2 ratio (at 5 × 10−5 mol L−1) in ammonium acetate/ammonium hydroxide buffer at pH 7.5 (Fig. 6). The ESI-MS spectrum (Fig. 6) shows an intense peak for (PC2 + H)+ at 540.15 m/z. A small peak at 179.05 m/z can be appreciated for (Cys-Gly + H)+, as well as the Cd pattern at 113.91 m/z. Moreover, other peaks related to the different complexes can be distinguished. Thus, peaks at 468.97 m/z for [Cd(Cys-Gly)2-H]+, at 652.04 m/z for [(CdPC2)-H]+ and at 1301.07 m/z for [Cd2(PC2)2-5H]+ are clearly identified. The obtained pattern of each of these complexes matches the isotope abundance of Cd. Thus, the presence of the main species detected by voltammetry is confirmed by MS experiments.
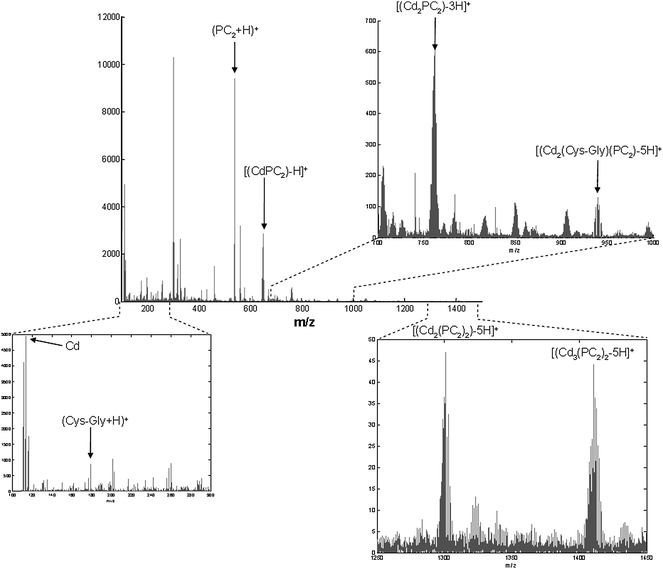 | ||
| Fig. 6 Positive ESI-MS spectrum for a 3:1:1 Cd/Cys-Gly/PC2 ratio (at 5 × 10−5 mol L−1) solution in 5 × 10−3 mol L−1 ammonium acetate/ammonium hydroxide buffer at pH 7.5. | ||
However, ESI-MS results also revealed the possible existence of [(Cd2(PC2)2)-3H]+ at 761.93 m/z and [(Cd3(PC2)2)-5H]+ at 1410.96 m/z, and also the presence of a mixed ligand Cd complex in the form of [(Cd2(Cys-Gly)(PC2)-5H]+ at 939.97 m/z, with a clear Cd pattern in all cases. This suggests that the existence of Cd2PC2, Cd3(PC2)2 and Cd2(Cys-Gly)(PC2) can occur, at least to some extent.
From a qualitative point of view the presence of these complexes is compatible with the voltammetric results described above, because components in voltammetry are related with electrodic processes that do not necessarily correspond to chemical species. Thus, the Cd3(PC2)2 complex should maintain the signal of Cd bound to two PC2 units through the thiol group and give a new signal related to the reduction of the third Cd bound to the carboxylate groups, which should appear at less negative potentials than the other, and that would be masked by the signal of Cys-Gly or Cd2+ in excess. A similar phenomenon could happen with the mixed complex that may co-exist as a transition step in the formation of more stable complexes with PC2. Another possible explanation would be the presence of very low concentrations of three species in solution (so that they would not be detected by voltammetry) but with a high sensitivity in ESI-MS (thus yielding noticeable signals).
4.2 Cd/Cys/PC3 system
For the study of the Cd/Cys/PC3 system, PC3 is added to Cd:Cys solutions at different concentration ratios: i) Cd2+ ions are initially complexed (partially or totally) with the smaller ligand (Fig. 7a and b); and ii) Cys is added to a solution where Cd2+ is present in either free form or complexed with PC3 (Fig. 7c). These examples are similar to those described in the previous system (Fig. 2a–c), and similar evolutions are observed.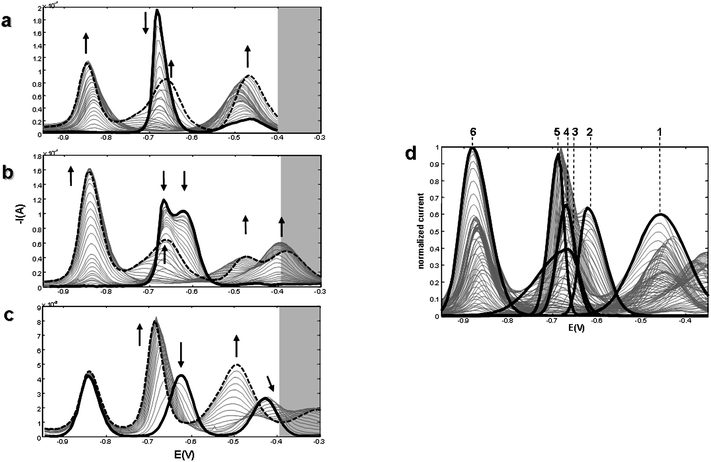 | ||
| Fig. 7 Evolution of DPP curves along PC3 or Cys additions to Cd/Cys or Cd/PC3 solutions respectively, in 0.1 mol L−1 TRIS-HNO3 buffer (pH 7.5): a) PC3 is added to a 1:2 Cd:Cys solution; b) PC3 is added to a 2:1 Cd:Cys solution; c) Cys is added to a 3:1 Cd:PC3 solution. Arrows indicate the signals tendency (— initial DPP curve; final DPP curve). d) Initial estimation of components for the MCR-ALS analysis. Numbers in figure indicate the components involved in the reduction process: Cys (1), Cd2+ (2), PC3 (3), Cd/Cys complexes (4 and 5) and Cd/PC3 complexes (6). Unitary concentration: 1 × 10−5 mol L−1. | ||
When PC3 is added to a 1:2 Cd2+:Cys solution (Fig. 7a), a drastic decrease in the Cd/Cys signal (at Ep = −0.69 V) simultaneously to the appearance, at more negative potentials, of the typical Cd/PC3 signal (at Ep = −0.88 V, Fig. 7a) is observed. Likewise, an increase of the anodic oxidation of Hg in the presence of free Cys takes place (at ca. −0.46 V). Throughout the titration, a remarkable movement in this signal is observed, mainly due to overlapping with the signal of the anodic oxidation of Hg in the presence of Cd/PC3 complex. At the end of the titration, the peak due to the excess of PC3 is observable at −0.66 V. As in the previous system, the ligand with more thiol groups (PC3) displaces Cd2+ of its complexes with the smaller chain ligand (Cys), giving new and more stable complexes.
When PC3 is added to a 2:1 Cd2+:Cys solution (Fig. 7b), free Cd2+ and Cd/Cys complexes are initially present, giving two overlapping peaks. As in the previous system, the successive additions of PC3 cause a decrease of both signals and an increase of the peak corresponding to Cd/PC3 complexes (at −0.88 V) together with the signals of the ligands.
Successive additions of Cys to a 1:3 Cd:PC3 solution (Fig. 7c) produces the depletion of the free Cd2+ and the formation of the successive Cys complexes (at −0.68 V), that coexist with the Cd/PC3 complex (Ep = −0.84 V). At the end of the titration, an intense and asymmetric peak related to the presence of both the new Cd/Cys complex and the previously existing Cd/PC3 complex is observed. Parallel to this, the characteristic signal related to the anodic oxidation of Hg in the presence of free Cys (Ep = −0.49 V) is observed, and the absence of the characteristic signal of free PC3 is remarkable.
The proper MCR-ALS resolution of data in Fig. 7a–c is obtained considering the six components shown in Fig. 7d as an initial estimate for ALS iterative optimization. Ligands in excess are assigned to components 1 and 3, Cys and PC3 respectively. Component 2 correlates to the reduction of free Cd2+ ions at −0.62 V. The reduction of Cd2+ bound to Cys is shown by the presence of component 4 and 5 (ML and ML2 respectively are expected in these type of titrations39). In the case of the Cd/PC3 complex only one component (6) was considered due to constant peak potential throughout the additions.
The constraints of non-negativity (for both concentrations and signals) and signal shape (for the voltammograms of all species) were applied. The MCR-ALS iterative process (preceded by the application of the shiftfit algorithm32 to components 1 and 3, signal correlated with each ligand in excess) yielded the unitary voltammograms as those assumed in Fig. 7d and the concentration profiles shown in Fig. 8a–c, with overall lacks of fit of 12.06, 12.96 and 8.92% respectively. Briefly, concentration profiles show that the successive addition of PC3 causes a decrease of the component assigned to Cd2+/Cys complexes (4/5) and free Cd2+(2), although, when present, the last clearly decreases more sharply (Fig. 8b). When data is plotted in front of added ligand-to-Cd ratios it is clear that the predominance of the Cd/PC3 complex (component 6) occurs at quite similar values. As expected, the addition of Cys to the solution where Cd2+ is partially complexed with PC3 (Fig. 8c) does not affect the concentration profile of Cd/PC3 complex (component 6), sequestering only the remaining free Cd2+ in solution and yielding components 4 and 5 (ML and ML2 respectively).
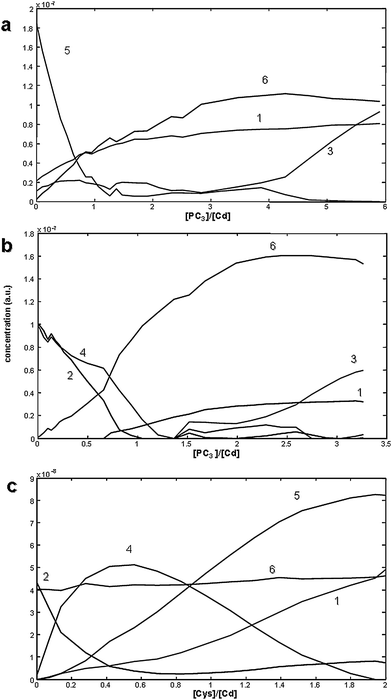 | ||
| Fig. 8 Respective concentration profiles obtained in the MCR-ALS decomposition of the data matrices shown in Fig. 7a–c. Applied constrains were non negativity and signal shape. The concentration profiles are plotted as function of the added ligand-to-total Cd2+ concentration ratios. Components are: Cys (1), Cd2+ (2), PC3 (3), Cd/Cys complexes (4 and 5) and Cd/PC3 complex (6). | ||
Positive ESI-MS data confirm the presence of Cd/PC3 complexes, ML (in majority) and M2L (poorly detected). Therefore, these results lead us to believe that component 6 is related to an ML (Cd/PC3) complex. No coherent Cd pattern of Cd/Cys complexes was detected, nor was a mixed ligand Cd2+ complex found (data not shown). Although Cd/Cys species were not detected in ESI-MS, their formation in solution is indisputable based on voltammetric data. The formation of a mixed ligand complex is plausible, although it must occur in very specific conditions and at low concentration levels. This system would be a favourable scenario for the occurrence of mixed ligand-metal complexes. Cys is the smallest PCn fragment containing a thiol group that yields stable complexes with Cd2+. Moreover, PC3 seems to have the adequate chain length to avoid bulk effects and, more importantly, it has three thiol groups. Considering that Cd2+ coordinates in a tetrahedral way, it would be probable that the Cd2+ would be bound to one PC3, through its three thiol groups, allowing for the possibility of Cd2+ coordination with a fourth thiol from Cys.
5. Conclusions
The work described herein shows the usefulness of voltammetry assisted by MCR-ALS for resolving complex mixtures of heavy metal ions and phytochelatin-related peptides. For this purpose, the comparison and, in some cases, the combination of datasets obtained separately for the different ligands appears to be crucial.For the particular systems here considered, the results are evidence that longer chain peptides containing more thiol groups easily displace shorter peptides from their metal complexes. Such displacements seem to occur quickly, since equilibrium is reached within the few minutes taken by every addition followed by solution stirring. Despite the fact that the systems were deliberately chosen to favour the formation of mixed ligand complexes to easily fulfil cadmium tetrahedral coordination, no voltammetric evidence of such species was found.
The complementary ESI-MS information confirms the presence in both systems of the majority species detected voltammetrically. In the case of the Cd2+-PC2-Cys-Gly mixture, ESI-MS detects also a few mixed ligand species. This could mean that: i) such species exist in solution in very low concentrations that are not detectable by voltammetry, ii) they are detected by voltammetry, but their signals are so close to these of majority species that they cannot be distinguished from them, or iii) they do not exist in solution, but are created during the ionisation process of ESI-MS.
As hypothesis i) appears to be the most probable one, the question is why mixed ligand complexes are not stabilised as an intermediate stage between the substitution of several short chain ligands by the first and the second longer chain ligand. This could be related to steric effects or to the tendency of some thiol peptides to join two long chain molecules with the binding of several metal ions. In either case, further experiments with complementary techniques would be required to resolve this question.
Finally, further voltammetric studies towards the competitive metal binding of different chain-length phytochelatins would be of great interest, along with examining how the chain-length influences the complexation process. This information could be very useful to genetically modify the plants to improve their phytoremediation efficiency.
Acknowledgements
The authors acknowledge financial support from the Spanish Ministerio de Ciencia e Innovación (projects CTQ2006-14385-C02-01 and CTQ2009-09471). Rui Gusmão also thanks the Spanish Ministerio de Ciencia e Innovación for his Ph.D. grant (BES-2007-15385). The authors are also grateful for the aid received from E. Chekmeneva. Last but not least, helpful discussions about the ESI-MS experiments with Laura Ortiz and Irene Vidal are also acknowledged.References
- G. Bertin and D. Averbeck, Biochimie, 2006, 88, 1549 CrossRef CAS.
- S. Chatterjee, S. Kundu, S. Sengupta and A. Bhattacharyya, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2009, 663, 22 CrossRef CAS.
- C. Cobbett and P. Goldsbrough, Annu. Rev. Plant Biol., 2002, 53, 159 CrossRef CAS.
- E. Grill, E. -L. Winnacker and H. H. Zenk, Science, 1985, 230, 674 CrossRef CAS.
- M. H. Zenk, Gene, 1996, 179, 21 CrossRef CAS.
- D. E. Salt and W. E. Rauser, Plant Physiol., 1995, 107, 1293 CAS.
- E. Pilon-Smits, Annu. Rev. Plant Biol., 2005, 56, 15 CrossRef CAS.
- I. Raskin and B. D. Ensley, Phytoremediation of ToxicMetals: Using Plants to Clean Up the Environment, Wiley, New York, 2000 Search PubMed.
- S. Cherian and M. M. Oliveira, Environ. Sci. Technol., 2005, 39, 9377 CrossRef CAS.
- S. L. Doty, New Phytol., 2008, 179, 318 Search PubMed.
- K. Hirata, N. Tsuji and K. Miyamoto, J. Biosci. Bioeng., 2005, 100, 593 CrossRef CAS.
- J. Szpunar, Analyst, 2000, 125, 963 RSC.
- E. Bramanti, D. Toncelli, E. Morelli, L. Lampugnani, R. Zamboni, K. E. Miller, J. Zemetra and A. D'Ulivo, J. Chromatogr., A, 2006, 1133, 195 CrossRef CAS.
- H. Chassaigne, V. Vacchina and R. Lobinski, Trends Anal. Chem., 2000, 19, 300 CrossRef CAS.
- T.-Y. Yen, J. A. Villa and J. G. DeWitt, J. Mass Spectrom., 1999, 34, 930 CrossRef CAS.
- A. P. Navaza, M. Montes-Bayo, D. L. LeDuc, N. Terry and A. Sanz-Medel, J. Mass Spectrom., 2006, 41, 323 CrossRef CAS.
- L. Chen, Y. Guo, L. Yanga and Q. Wang, J. Anal. At. Spectrom., 2007, 22, 1403 RSC.
- R. K. Mehra, V. R. Kodati and R. Abdullah, Biochem. Biophys. Res. Commun., 1995, 215, 730 CrossRef CAS.
- B. J. Fuhr and D. L. Rabenstein, J. Am. Chem. Soc., 1973, 95, 6944 CrossRef CAS.
- G. Scarano and E. Morelli, Electroanalysis, 1998, 10, 39 CrossRef CAS.
- V. Supalkova, D. Huska, V. Diopan, P. Hanustiak, O. Zitka, K. Stejskal, J. Baloun, J. Pikula, L. Havel, J. Zehnalek, V. Adam, L. Trnkova, M. Beklova and R. Kizek, Sensors, 2007, 7, 932 CrossRef CAS.
- V. Dorĉák and I. Ŝestáková, Bioelectrochemistry, 2006, 68, 14 CrossRef CAS.
- P. C. White, N. S. Lawrence, J. Davis and R. G. Compton, Electroanalysis, 2002, 14, 89 CrossRef CAS.
- M. Heyrovský, P. Mader, S. Vavricka, V. Veselá and M. Fedurco, J. Electroanal. Chem., 1997, 430, 103 CrossRef CAS.
- M. Mladenov, V. Mircesk, I. Gjorgoski and B. Jordanosk, Bioelectrochemistry, 2004, 65, 69 CrossRef CAS.
- M. Yamamoto, T. Charoenraks, H. Pan-Hou, A. Nakano, A. Apilux and M. Tabata, Chemosphere, 2007, 69, 534 CrossRef CAS.
- M. Esteban, C. Ariño, J. M. Díaz-Cruz and R. Tauler, Trends Anal. Chem., 2000, 19, 49 CrossRef CAS.
- M. Esteban, C. Ariño and J. M. Díaz-Cruz, Trends Anal.Chem., 2006, 25, 86 CrossRef CAS.
- M. Esteban, C. Ariño and J. M. Díaz-Cruz, Crit. Rev. Anal. Chem., 2006, 36, 295 CrossRef CAS.
- Y. Ni and S. Kokot, Anal. Chim. Acta, 2008, 626, 30.
- A. de Juan and R. Tauler, Crit. Rev. Anal. Chem., 2006, 36, 163 CrossRef CAS.
- A. Alberich, C. Ariño, J. M. Díaz-Cruz and M. Esteban, Analyst, 2008, 133, 470 RSC.
- J. M. Díaz-Cruz, J. Agulló, M. S. Díaz-Cruz, C. Ariño and M. Esteban, Analyst, 2001, 126, 371 RSC.
- B. H. Cruz, J. M. Díaz-Cruz, C. Ariño, M. Esteban and R. Tauler, Analyst, 2002, 127, 401 RSC.
- B. H. Cruz, J. M. Díaz-Cruz, I. Ŝestáková, J. Velek, C. Ariño and M. Esteban, J. Electroanal. Chem., 2002, 520, 111 CrossRef CAS.
- B. H. Cruz, J. M. Díaz-Cruz, C. Ariño and M. Esteban, Environ. Sci. Technol., 2005, 39, 778 CrossRef CAS.
- E. Checkmeneva, R. Prohens, J. M. Díaz-Cruz, C. Ariño and M. Esteban, Environ. Sci. Technol., 2008, 42, 2860 CrossRef.
- A. Alberich, C. Ariño, J. M. Díaz-Cruz and M. Esteban, Anal. Chim Acta, 2007, 584, 403 CrossRef CAS.
- R. Gusmão, C. Ariño, J. M. Díaz-Cruz and M. Esteban, Anal. Bioanal. Chem., 2009, 394, 1137 CrossRef CAS.
- The MathWorks ( 2006) MATLAB version 7.3.0.267. The Math-Works, Natick Search PubMed.
- A. Alberich, J. M. Díaz-Cruz, C. Ariño and M. Esteban, Analyst, 2008, 133, 112 RSC.
| This journal is © The Royal Society of Chemistry 2010 |