Dual contactless conductivity and amperometric detection on hybrid PDMS/glass electrophoresis microchips†
Mercedes
Vázquez
ab,
Celeste
Frankenfeld
b,
Wendell K. Tomazelli
Coltro
bc,
Emanuel
Carrilho
c,
Dermot
Diamond
a and
Susan M.
Lunte
*b
aCentre for Bioanalytical Sciences, National Centre for Sensor Research, Dublin City University, Glasnevin, Dublin 9, Dublin, Ireland
bRalph N. Adams Institute for Bioanalytical Chemistry, MRB, The University of Kansas, Lawrence, KS 66047-1620, USA. E-mail: slunte@ku.edu; Fax: +1 785 864 1916; Tel: +1 785 864 3811
cInstituto de Química de São Carlos, Universidade de São Paulo, São Carlos-SP, Brazil
First published on 10th November 2009
Abstract
A new approach for the integration of dual contactless conductivity and amperometric detection with an electrophoresis microchip system is presented. The PDMS layer with the embedded channels was reversibly sealed to a thin glass substrate (400 μm), on top of which a palladium electrode had been previously fabricated enabling end-channel amperometric detection. The thin glass substrate served also as a physical wall between the separation channel and the sensing copper electrodes for contactless conductivity detection. The latter were not integrated in the microfluidic device, but fabricated on an independent plastic substrate allowing a simpler and more cost-effective fabrication of the chip. PDMS/glass chips with merely contactless conductivity detection were first characterized in terms of sensitivity, efficiency and reproducibility. The separation efficiency of this system was found to be similar or slightly superior to other systems reported in the literature. The simultaneous determination of ionic and electroactive species was illustrated by the separation of peroxynitrite degradation products, i.e. NO3− (non-electroactive) and NO2− (electroactive), using hybrid PDMS/glass chips with dual contactless conductivity and amperometric detection. While both ions were detected by contactless conductivity detection with good efficiency, NO2− was also simultaneously detected amperometrically with a significant enhancement in sensitivity compared to contactless conductivity detection.
1. Introduction
In recent years, much of the research in the area of electrophoresis microchips has been focused in the search for alternative detection systems offering an improved performance and an ease of integration and miniaturization.1–3 A simple search in the literature shows that laser-induced fluorescence (LIF) and amperometric detection2,3 have been traditionally the choices in this area due to their great sensitivity and selectivity. However, those detection modes are not universal. In the case of LIF, analytes must fluoresce or be previously derivatized with fluorophores in order to be detected. Likewise, analytes must be electrochemically active for amperometric detection. Another electrochemical technique, namely, capacitively coupled contactless conductivity detection (C4D), has gained much popularity as on-chip detection in electrophoresis microchips since its introduction on conventional capillary electrophoresis (CE) in 1998.4,5 Conductivity detection is a universal detection technique that involves the measurement of the differences in the conductivity of the electrolyte solution compared to that of the analyte zones. The integration of C4D with the microfluidic device is rather simple due to the lack of physical contact of the detection electrodes with the electrolyte solution. Additionally, ease of miniaturization and low-cost instrumentation have played an important role in the current success of C4D in the electrophoresis microchip field.New trends in microfluidics, however, are focused on the coupling of two detection techniques in a single electrophoresis microchip rather than on the improvement of individual detection modes. Combination of the inherent properties of individual detection modes in a dual detection system has become a powerful tool to greatly enhance sample characterization and widen the range of application. However, not many publications have explored this concept to date. Obviously, the coupling of two different detection techniques in a single microfluidic device is not trivial and many parameters have to be considered, such as compatibility of both detection modes and ease of their integration with the microchip. Dual detection systems coupling fluorescence with light-scattering,6 and amperometry with LIF,7 C4D8 and electrochemiluminescence (ECL)9 have been reported so far. Additionally, a dual-electrode electrochemical detection was also introduced.10
In this work, a new approach for the integration of an electrophoresis microchip with dual C4D and amperometric detection is introduced. The coupling of C4D and amperometric detection has been reported earlier in connection with commercially available glass microchips.8 To the best of our knowledge, this is the first time that the successful integration of both detection techniques with an in-house hybrid PDMS/glass chip is reported. A description of the analytical characteristics of this new system and its advantages compared to the system presented by Wang et al.8 is presented in this report. Additionally, the hybrid PDMS/glass microchip with dual C4D and amperometric detection was used in the determination of peroxynitrite (ONOO−) degradation products. Peroxynitrite is a reactive oxidative species that forms within the body from the reaction of the free radicals nitric oxide (˙NO) and superoxide (O2˙−).11 Once formed, ONOO− rapidly decomposes to species that can indiscriminately oxidize and nitrate surrounding tissue. When the production of ONOO− is balanced by the scavenging actions of endogenous antioxidants, or when it is generated by the immune system to attack foreign microbes, the break-down of ONOO− is not harmful. However, in instances such as inflammation, excess ONOO− and its decomposition products can denature DNA,12 destroy lipid membranes13,14 and alter protein function.15,16 These destructive effects of ONOO− have been implicated in the development of atherosclerosis,17–19 diabetes,19,20 and neurodegenerative21 and autoimmune diseases.22,23
The precise degradation mechanism for ONOO− is still under debate,24–27 although it is well known that peroxynitrite degrades rapidly into its main metabolites, i.e., nitrate (NO3−) and nitrite (NO2−). A CE method for the separation and direct UV detection of peroxynitrite, nitrate and nitrite has been published elsewhere.28 However, direct detection of peroxynitrite is still very challenging due to its extremely short lifetime, and most analytical methods are based on the detection of its degradation products.26 Here, we present a new method for the separation and detection of NO3− and NO2− resulting from degradation of peroxynitrite using a hybrid PDMS/glass chip with C4D and amperometric detection. Both anions can be detected by C4D; however, only nitrite can be detected by amperometry as it is the only electrochemically active species from the two anions. We found that the coupling of C4D and amperometric detection allowed a more comprehensive analysis of peroxynitrite samples.
2. Experimental
2.1. Materials and reagents
The following chemicals and materials were used as received: SU-8 10 negative photoresist and Nano SU-8 developer (MicroChem Corp., Newton, MA); AZ 1518 positive photoresist and 300 MIF developer (AZ Resist, Somerville, NJ). Silicon wafers were obtained from Silicon, Inc. (Boise, ID). Sylgard 184 elastomer and curing agent were obtained from Ellsworth Adhesives (Germantown, WI). Silver colloidal paste (Ted Pella, Inc., Redding, CA), JB Weld epoxy (JB Weld, Sulfur Springs, TX), buffer oxide etch (BOE) (J.T. Baker), copper etching solution (Radio Shack, Lawrence, KS) and copper wire (22 gauge, Westlake Hardware, Lawrence, KS) were used as received. Pd (99.95% purity) and Ti (99.97% purity) targets (2 in diameter × 0.125 in thickness) were purchased from Kurt J. Lesker Co., Clairton, PA. Wax paper sheets were obtained from Ferragini (Sao Carlos-SP, Brazil). 2-(N-morpholino)ethanesulfonic acid (MES), DL-histidine, DL-lactic acid and tetradecyltrimethylammonium hydroxide (TTAOH) were obtained from Sigma (St. Louis, MO). Acetone, isopropyl alcohol, H2O2 (30%), H2SO4, HNO3, NaOH and HCl were obtained from Fisher Scientific (Fair Lawn, NJ). Peroxynitrite was freshly synthesized in-house following the reaction of acidified nitrite and hydrogen peroxide.29 All other chemicals were analytical-grade reagents. Ultrapure water (resistivity 18.2 MΩ cm) was used to prepare all solutions, which were filtered through 0.22 μm filters prior to use.2.2. Electrode fabrication
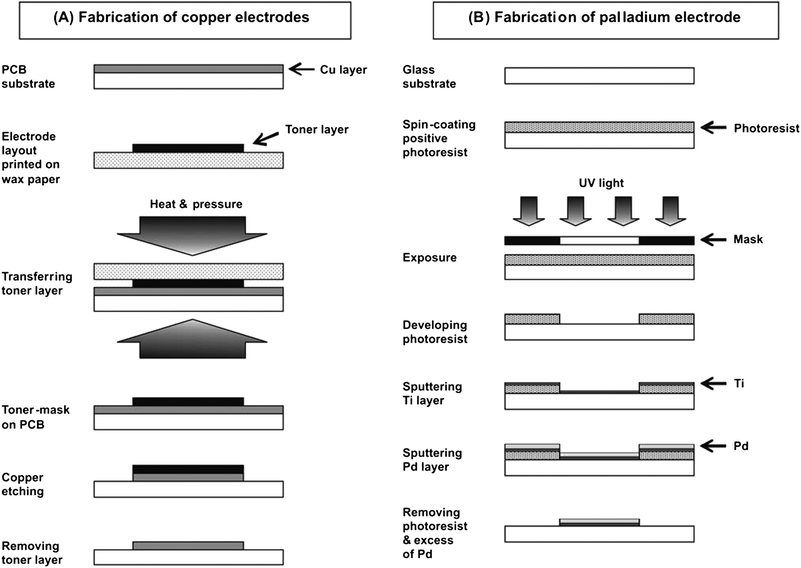 | ||
| Fig. 1 Fabrication of copper electrodes for C4D detection (A) and palladium electrode for amperometric detection (B). | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O2, 7:3) and then dipped in BOE for 40 s. A thin layer of the positive photoresist AZ 1518 was then applied on top of the glass substrate by using a spin-coater (Brewer Science, Rolla, MO) at 2000 rpm for 40 s (pre-baking step, 1 min at 100 °C). Afterwards, a dark field mask (the electrode pattern transparent, the rest of the mask black) was placed on top of the photoresist layer and the glass substrate was exposed to UV light (Optical Associates Autoflood 1000 UV floodsource, Milpitas, CA) for 10 s. The photoresist was then developed in 300 MIF developing solution for 45 s and post-baked at 100 °C for 1 min. Next, the glass substrate with the developed pattern of photoresist was placed in the thin layer deposition system and a Ti layer (200 Å) was first deposited in order to improve the adhesion of the Pd layer (2000 Å). Finally, the photoresist layer was removed by immersion of the glass substrate in an acetone bath. As a result, the Ti and Pd layers deposited on top of the photoresist were lifted off revealing the pattern of the Pd electrode (40 μm width, 30 mm length). To enable electrical contact between the Pd electrode and the instrument, a copper wire was connected to the Pd electrode by using silver colloidal paste.
:H2O2, 7:3) and then dipped in BOE for 40 s. A thin layer of the positive photoresist AZ 1518 was then applied on top of the glass substrate by using a spin-coater (Brewer Science, Rolla, MO) at 2000 rpm for 40 s (pre-baking step, 1 min at 100 °C). Afterwards, a dark field mask (the electrode pattern transparent, the rest of the mask black) was placed on top of the photoresist layer and the glass substrate was exposed to UV light (Optical Associates Autoflood 1000 UV floodsource, Milpitas, CA) for 10 s. The photoresist was then developed in 300 MIF developing solution for 45 s and post-baked at 100 °C for 1 min. Next, the glass substrate with the developed pattern of photoresist was placed in the thin layer deposition system and a Ti layer (200 Å) was first deposited in order to improve the adhesion of the Pd layer (2000 Å). Finally, the photoresist layer was removed by immersion of the glass substrate in an acetone bath. As a result, the Ti and Pd layers deposited on top of the photoresist were lifted off revealing the pattern of the Pd electrode (40 μm width, 30 mm length). To enable electrical contact between the Pd electrode and the instrument, a copper wire was connected to the Pd electrode by using silver colloidal paste.
2.3. PDMS/glass chip fabrication
The PDMS/glass chips fabricated were based on the standard configuration used in electrophoresis microchips, i.e., a simple cross configuration of the injection and the separation channels. First, a layer of PDMS containing the two channels in a cross configuration was fabricated by soft lithography.34,35 Briefly, the channel patterns printed on high-resolution transparency film (dark field mask) by Laser Graphics Co. (Lawrence, KS) were transferred to a silicon wafer. In order to do that, a thin layer of the negative photoresist SU-8 10 was previously spin-coated on top of a clean silicon wafer. Then, the dark field mask was placed on top of the photoresist layer and the wafer was exposed to UV light for 3 min. Next, the photoresist was developed in Nano SU-8 developer revealing a positive pattern of the channels. The height of the channels was then measured using a profilometer (Tencor Alphastep 200, Mountain View, CA). Afterwards, a mixture of PDMS and curing agent (Sylgard 184) in a proportion of 10:1 was casted on top of the silicon wafer and cured for at least 1 h at 80 °C. The PDMS was then peeled off the silicon wafer revealing a pattern of negative channels on the PDMS layer. Sample and buffer reservoirs were made in the PDMS layer with a hole punch. Finally, the PDMS layer was reversibly sealed to a thin glass substrate (0.4 ± 0.1 mm thickness) in order to produce a hybrid PDMS/glass chip. To enable C4D, the PDMS/glass chip was then securely fixed to a plastic substrate (PCB) containing the copper electrodes (section 2.2.1) (Fig. 2A). The channel dimensions of this chip (70 × 30 mm) were the following: the injection and separation channels were 20 mm and 50 mm long, respectively, 150 μm wide and 15 μm deep.
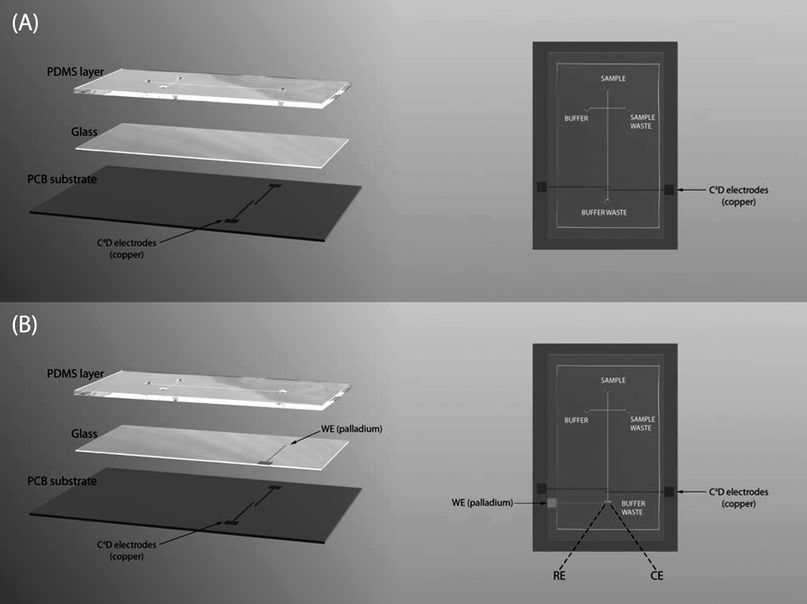 | ||
| Fig. 2 Layout of the hybrid PDMS/glass chip with C4D (A) and C4D coupled in line with amperometric detection (B). Chip dimensions: (A) 70 × 30 mm and (B) 75 × 30 mm. Channel dimensions: (A) 20 mm-long injection channel, 50 mm-long separation channel, 150 μm wide and 15 μm deep; (B) 20 mm-long injection channel, 57 mm-long separation channel, 30 μm wide and 12 μm deep. WE: working electrode; RE: reference electrode; CE: counter electrode. | ||
The design of the PDMS/glass chip described above was then slightly modified in order to integrate both detection modes, i.e., C4D and amperometric detection, in the same chip. In this case, a glass substrate (0.4 ± 0.1 mm thickness) containing a Pd electrode (section 2.2.2) was reversibly sealed to the PDMS layer with the Pd electrode facing the PDMS layer (Fig. 2B). The alignment of the Pd electrode at 5–20 μm from the end of the separation channel was done with the help of an optical microscope. The channel dimensions of the chip (75 × 30 mm) were the following: injection and separation channels were 20 mm and 57 mm long, respectively, 30 μm wide and 12 μm deep.
2.4. Electrophoresis procedure
The buffer employed in the separation of the selected cations (potassium, sodium and lithium) and anions (chloride, nitrate and perchlorate) was 20 mM MES/His at pH 6.1. Stock solutions of the target ions (10 mM) were prepared from the corresponding chloride salts (for cations) and sodium salts (for anions) in the run buffer, and stored at 4 °C. Subsequent dilutions of the stock solutions were done either in water or in the run buffer.Stock solutions of NO2− and NO3− at 10 mM concentration were freshly prepared in water each week from their corresponding sodium salts, and kept at 4 °C between measurements. Different run buffers containing tetradecyltrimethylammonium hydroxide (TTAOH) as electroosmotic flow (EOF) modifier were tested for the analysis of standards containing both anions dissolved in water. Successful separation of NO2− and NO3− was finally accomplished by using 30 mM lactic acid–0.75 mM TTAOH at pH 3.5 as run buffer.
Peroxynitrite (ONOO−) was freshly synthesized in-house prior to analysis following the acidified nitrite and hydrogen peroxide reaction.29 After synthesis, the concentration of peroxynitrite was calculated based on the UV absorbance at 302 nm at which maximum absorbance for peroxynitrite occurs. The resulting reaction product had a concentration in peroxynitrite of ca. 60 mM. The separation of NO2− and NO3− resulting from the degradation of peroxynitrite was achieved by using 30 mM lactic acid–0.75 mM TTAOH at pH 3.5.
A high voltage power supply (Jenway, Essex, England) with 4 channels (0 kV ± 5 kV) and controlled by a computer was used for the electrokinetic injection and flow control. Channels were preconditioned with buffer solution for 10 min prior to use. Gated injection was employed for all electrokinetic injections due to its flexibility regarding sample plug size and its reproducibility relative to unpinched injection. Thus, a high voltage was applied to the buffer reservoir in order to fill the injection channel, and a fraction of that voltage (0.67) was applied to the sample reservoir while keeping sample and buffer waste reservoirs grounded. Sample injection was then performed by floating the voltage at the buffer reservoir for 1 s, which drove the sample plug into the separation channel. The voltage at the buffer reservoir was then resumed to allow sample separation. All the experiments were performed at room temperature (23 ± 2 °C).
2.5. Capacitively coupled contactless conductivity detection (C4D)
Contactless conductivity detection was performed by using an in-house C4D system fabricated in the Instituto de Química de São Carlos (University of São Paulo, Brazil) as described elsewhere.36,37 In order to prevent external interferences, it was necessary to keep the PDMS/glass chip inside a shielding box during measurements. A function generator model 4003A from BK Precision Co. (Yorba Linda, CA) was used to generate the excitation sinusoidal signal (frequency, 450–500 kHz; excitation voltage, 10–15 Vpeak-to-peak) applied to the actuator electrode, and the resulting signal was then measured at the pick-up electrode (fabrication of the sensing electrodes described in section 2.2.1). An Epsilon potentiostat (Bioanalytical Systems, Lafayette, IN) was used for conversion of the analog signal into a digital signal.2.6. Amperometric detection
A three-electrode system was used for amperometric detection. The working electrode was a Pd electrode (fabrication described in section 2.2.2) placed at the end of the separation channel (Fig. 2B). A Pt wire was used as auxiliary electrode and the reference electrode was a Ag|AgCl|KCl (3 M) electrode. The auxiliary and reference electrodes were embedded in the buffer waste reservoir at the end of the separation channel, i.e., detection reservoir. Amperometric detection was done by means of an Epsilon potentiostat (Bioanalytical Systems, Lafayette, IN) after applying 850 mV to the working electrode.3. Results and discussion
3.1. Validation of the PDMS/glass chips with C4D
Mixtures of small inorganic cations and anions were used to evaluate the analytical performance of hybrid PDMS/glass microchips with contactless conductivity detection. For these studies, the chip design described in section 2.3 (Fig. 2A) having a separation channel of ca. 35 mm effective length (from the cross intersection to the mid-point between the C4D electrodes), 150 μm width and 15 μm depth, was used. The sensing copper electrodes were not integrated in the microfluidic device, but fabricated on an independent plastic substrate following the procedure described in section 2.2.1 (Fig. 1A). This allowed a simpler and more cost-effective fabrication of the chip, which also represents an advantage for chip mass production. Furthermore, the replacement of chips was easily done when needed as the PDMS/glass chip was simply secured with tape to the plastic substrate containing the sensing electrodes.Mixtures of sodium, potassium and lithium cations dissolved in water at equimolar concentrations were injected by applying 0.8 kV for 1 s along the injection channel. The electrophoretic separation of the target cations was achieved by applying 1.2 kV along the separation channel using 20 mM MES/His, pH 6.1, as run buffer. As can be seen in Fig. 3A, separation of the three cations at 500 μM (each) was achieved within 40 s with very reproducible migration times (RSD < 0.96%, n = 4). Additionally, well defined and resolved peaks were obtained under these conditions. The plate number per meter (N/m) calculated for those peaks by the 5-sigma method was 4.9 × 104 for potassium, 6.6 × 104 for sodium, and 9.5 × 104 for lithium. Thus, the separation efficiency of this system is similar or slightly superior to other systems reported in the literature (4.6–5.4 × 104 plates/m for sodium in ref. 38 and 4.3 × 104 plates/m for potassium in ref. 39). Limits of detection of 31 μM, 35 μM and 50 μM were estimated (signal to noise ratio = 3) for potassium, sodium and lithium, respectively, based on the response observed for a mixture containing the target cations at 50 μM (see Fig. S-1 in the ESI†). Lower detection limits in the range of 11 μM to 25 μM were found upon increasing the injection time from 1 s to 3 s (see Fig. S-2 in the ESI†). One of the possible causes for the slightly lower sensitivity of our system compared to others reported in the literature using polyester-toner,37 PMMA38,40 or glass39 chips is related to the thickness of the layer separating the sensing electrodes (copper) from the sample/electrolyte flowing in the microchannel. Specifically, a glass substrate of ca. 400 μm was used to produce the hybrid PDMS/glass chip employed in this work (Fig. 2A). This glass substrate served also as a physical separation between the sensing electrodes and the microchannel and, thus, the distance between sensing electrodes and microchannel was larger than in those other devices (400 μm versus 100 μm in ref. 37, 125 μm in ref. 38,10–15 μm in ref. 39, and 100 μm in ref. 40). Therefore, in order to increase the sensitivity using hybrid PDMS/glass chips with C4D, the use of a glass substrate with thickness ≤ 200 μm33 and/or the application of a higher excitation voltage33,38,40 is recommended. The former can be simply attained by using commercially available microscope cover slides of the appropriate width and length.
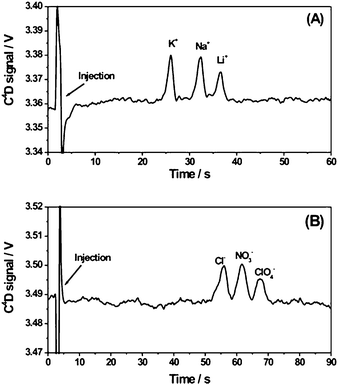 | ||
| Fig. 3 Electrophoretic separation of a 500 μM equimolar mixture of potassium, sodium and lithium chloride dissolved in water (A) and a 500 μM equimolar mixture of chloride, nitrate and perchlorate dissolved in run buffer (B). Separation parameters: run buffer, 20 mM MES/His, pH 6.1; injection voltage, 0.8 kV (A) and −0.8 kV (B); separation voltage, 1.2 kV (A) and −1.2 kV (B); injection time, 1 s; separation channel effective length, ca. 35 mm. C4D parameters: frequency, 500 kHz; excitation voltage, 10 Vpeak-to-peak. | ||
Additionally, equimolar mixtures of chloride, nitrate and perchlorate anions dissolved in run buffer (20 mM MES/His, pH 6.1) were injected at −0.8 kV (injection time, 1 s), and subsequently separated by applying −1.2 kV along the separation channel. As can be seen in Fig. 3B, separation of the three anions at 500 μM (each) was achieved within 65 s (RSD < 0.7% for three consecutive injections). Once more, the sensitivity of this system for the anions tested was found to be lower than in other systems reported earlier.40 Limits of detection of 79 μM, 67 μM and 77 μM were estimated (signal to noise ratio = 3) for chloride, nitrate and perchlorate, respectively, based on the response observed for a mixture containing the target anions at 100 μM (see Fig. S-3 in the ESI†). Wider peaks were observed for the anions compared to the cations tested (Fig. 3A), which could be caused by a larger contribution of diffusional band broadening as a result of longer migration times.40 However, the resolution was still good enough (Rs = 1.2) to allow a satisfactory separation of the target anions with rather good efficiencies (5.5 × 104 plates/m for chloride, 7.9 × 104 plates/m for nitrate and 13.0 × 104 plates/m for perchlorate).
3.2. Analysis of peroxynitrite by dual C4D and amperometric detection
Dual electrochemical detection was successfully applied in the analysis of peroxynitrite degradation products, i.e., NO2− and NO3−, by using the chip design depicted in Fig. 2B. While both ions can be detected by C4D, only electroactive species, i.e., nitrite (NO2−), can be detected by amperometric detection at much lower concentrations.2 Microchips with longer and narrower channels than in the previous section were used in order to improve the separation efficiency and resolution for conductivity detection. Thus, the separation channel had an effective length of ca. 42 mm for the C4D (from the cross intersection to the mid-point between the C4D electrodes), and ca. 47 mm for the amperometric detection (from the cross intersection to the palladium electrode), a width of 30 μm and a depth of 12 μm.The fabrication of the palladium electrode for amperometric detection (section 2.2.2) and its integration in the hybrid PDMS/glass chip was rather straightforward, and made the construction of a special chip/electrode holder unnecessary. The latter represents a clear advantage as compared to other systems using screen-printing electrodes as working electrodes for amperometric detection.8 Moreover, replacement of the PDMS/glass chips in the event of any channel fouling was easily done by peeling off the used PDMS layer from the glass containing the working electrode and reversibly sealing a new PDMS layer.
The selection of the background electrolyte (BGE) for this application was not trivial. BGEs of low conductivity are typically used for conductivity detection because they enhance sensitivity by maximizing the difference in conductivity between the analyte zone and the carrier electrolyte.41 However, buffers commonly used for amperometric detection are highly conducting and, thus, generate significant baseline noise when used for conductivity detection, with a consequent loss of sensitivity. The separation of NO2− and NO3− further complicates buffer selection due to their similar electrophoretic mobilities.42
Optimization of the separation conditions for the determination of ONOO− degradation products was carried out by using standards. A mixture of nitrate and nitrite anions dissolved in water was injected at −1.0 kV (injection time, 1 s) and then separated by applying −1.5 kV along the separation channel. In order to reverse the EOF, a cationic surfactant was added to the run buffer. Considering that TTAOH had been previously used for the separation of nitrate and nitrite by CE with contact conductivity detection,43 that was the EOF modifier of choice. As shown in Fig. 4, the separation of nitrate (1 mM) and nitrite (5 mM) anions was finally achieved in less than 50 s (RSD < 1.2% for three consecutive injections) by using 30 mM lactic acid–0.75 mM TTAOH, pH 3.5, as run buffer. This BGE exhibited the appropriate conductivity and ionic strength to allow a good separation by C4D (Fig. 4A). The effect of pH on the separation was proved to be very significant. No satisfactory resolution was achieved by using the same buffer composition at lower pH (pH 2.5 vs. pH 3.5). Therefore, the pH, near the pKa of NO2−, was essential for the separation of both ions by microchip electrophoresis with C4D. However, a better signal-to-noise ratio was observed for the NO2− peak by amperometric detection (Fig. 4B), together with flat baselines and low noise levels. Limits of detection were estimated (signal to noise ratio = 3) based on the response observed for a mixture containing 1 mM nitrite and 0.2 mM nitrate. Limits of detection found for nitrite with C4D (Fig. 4A) and amperometric detection (Fig. 4B) were 308 μM and 20 μM, respectively. The detection limit estimated for nitrate based only on C4D was 67 μM, in good agreement with the value previously calculated in section 3.1. The increased sensitivity shown by amperometric detection as compared to C4D is consistent with the LOD values reported in the literature for electrophoresis microchips.2 Good separation efficiency was found for C4D with 3.8 × 104 plates/m for nitrite and 4.5 × 104 plates/m for nitrate. It should be noted that the effective length of the separation channel for amperometric detection (ca. 47 mm) was longer than for C4D (ca. 42 mm) what resulted in a shift of the nitrite peak towards longer migration times for amperometric detection (Fig. 4). This peak shift can be prevented by transforming the time-based x-axis electropherograms obtained for both detectors into the corresponding effective mobility-scaled x-axis,44 as shown in the ESI†.
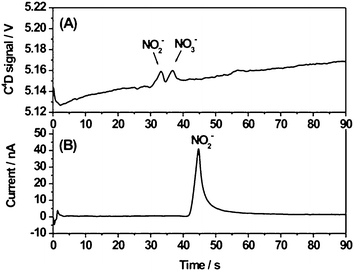 | ||
| Fig. 4 Electrophoretic separation of a mixture of 5 mM NO2− and 1 mM NO3− in water detected simultaneously by: A) C4D (frequency, 450 kHz; excitation voltage, 15 Vpeak-to-peak; effective length, ca. 42 mm); B) amperometric detection (voltage applied to the working electrode, 850 mV; effective length, ca. 47 mm). Separation parameters: run buffer, 30 mM lactic acid–0.75 mM TTAOH, pH 3.5; injection voltage, −1.0 kV; separation voltage, −1.5 kV; injection time, 1 s. | ||
Finally, the peroxynitrite sample synthesized in-house (ca. 60 mM) was analyzed by using the separation conditions optimized for nitrate and nitrite standards. Peroxynitrite is very unstable at pH lower than 12.28 Therefore, at pH 3.5, peroxynitrite degrades rapidly into its main metabolites, i.e., NO2− and NO3−. As can be seen in Fig. 5, the concentrations of NO2− and NO3− generated from the peroxynitrite degradation differed significantly. While a well-defined peak was observed for NO3−, both detectors showed distorted peaks for NO2− indicating nitrite overloading. However, further dilution of the peroxynitrite sample was not attempted as that could have prevented the satisfactory determination of nitrate by C4D. As shown in Fig. 4, a shift of the nitrite peak towards longer migration times is also observed in Fig. 5 for amperometric detection (effective length, ca. 47 mm) compared to C4D (effective length, ca. 42 mm). See the corresponding effective mobility-scaled electropherograms in the ESI†.
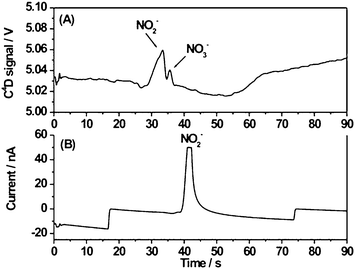 | ||
| Fig. 5 Electrophoretic separation of NO2− and NO3− resulting from the degradation of peroxynitrite (ca. 60 mM) detected simultaneously by: A) C4D (frequency, 450 kHz; excitation voltage, 15 Vpeak-to-peak; effective length, ca. 42 mm); B) amperometric detection (voltage applied to the working electrode, 850 mV; effective length, ca. 47 mm). Separation parameters: run buffer, 30 mM lactic acid–0.75 mM TTAOH, pH 3.5; injection voltage, −1.0 kV; separation voltage, −1.5 kV; injection time, 1 s. | ||
The overall aim of this research was to demonstrate the suitability of the proposed microchip-based electrophoretic system for the detection of analytes with very different electrochemical properties in real samples, and thus, no attempt was made to accurately quantify the concentrations of NO2− and NO3− resulting from the degradation of peroxynitrite. However, it was still possible to obtain estimations of those by extrapolation of the calibration curves calculated for the nitrate and nitrite standards with the C4D detector. These were 1.1 mM for NO3− (calibration curve correlation coefficient, R = 0.9299) and 22.2 mM for NO2− (R = 0.9393), respectively.
4. Conclusions
The application of hybrid PDMS/glass microchips with dual C4D and amperometric detection in the simultaneous determination of ionic and electroactive species was demonstrated. Electrophoretic separation of the main metabolites resulting from the degradation of peroxynitrite at low pH, i.e., NO2− (electroactive) and NO3− (non-electroactive), was achieved within 50 s by using 30 mM lactic acid–0.75 mM TTAOH, pH 3.5, as run buffer. The simultaneous use of both detectors provides complimentary information arising from the combination of the individual characteristics offered by each detection mode. Thus, while both ions were detected by C4D with good efficiency, nitrite was also simultaneously detected amperometrically with a significant enhancement in sensitivity compared to C4D. Additionally, the microfluidic platform proposed here has intrinsic advantages associated with the procedure followed in the fabrication of the microchip and the sensing electrodes. First, the method used to fabricate the very robust C4D electrodes is extremely low-cost, simple and versatile. Second, the fabrication of the C4D electrodes in an independent substrate allows an easy integration of the microchip with the C4D system and a straightforward replacement of the microchip when needed. Additionally, this simple setup offers great flexibility in the positioning of the detection point along the separation channel, which represents an advantage during the optimization of the separation method. Third, the integration of the working electrode for amperometric detection in the microchip made the construction of a chip and/or electrode holder unnecessary and allowed easier replacement of the microchip by simple substitution of the used PDMS layer. Therefore, this represents a very promising approach to the construction of a simple and low-cost microfluidic system incorporating a dual detection scheme that can provide more reliable analytical performance than it is possible using either of the individual detectors on their own.Acknowledgements
Financial support from the Industrial Development Authority, Ireland, (IDA project # 116294), Science Foundation Ireland (SFI Walton fellowship award 04/W4/B640 to Prof. Susan M. Lunte), the Coordenadoria de Aperfeiçoamento de Pessoal de Ensino Superior (CAPES) (Proc. No. BEX0706/02-1), the American Heart Association (Heartland Affiliation Predoctoral Fellowship), and the National Institutes of Health (Grant No. NS049229) is gratefully acknowledged. We would also like to thank Manuel Martínez for the drawings in Fig. 2.References
- P. S. Dittrich, K. Tachikawa and A. Manz, Anal. Chem., 2006, 78, 3887 CrossRef CAS.
- W. R. Vandaveer IV, S. A. Pasas-Farmer, D. J. Fischer, C. N. Frankenfeld and S. M. Lunte, Electrophoresis, 2004, 25, 3528 CrossRef.
- K. Uchiyama, H. Nakajima and T. Hobo, Anal. Bioanal. Chem., 2004, 379, 375 CrossRef CAS.
- A. J. Zemann, E. Schnell, D. Volgger and G. K. Bonn, Anal. Chem., 1998, 70, 563 CrossRef CAS.
- J. A. Fracassi da Silva and C. L. do Lago, Anal. Chem., 1998, 70, 4339 CrossRef.
- D. P. Schrum, C. T. Culbertson, S. C. Jacobson and J. M. Ramsey, Anal. Chem., 1999, 71, 4173 CrossRef CAS.
- J. A. Lapos, D. P. Manica and A. G. Ewing, Anal. Chem., 2002, 74, 3348 CrossRef.
- J. Wang and M. Pumera, Anal. Chem., 2002, 74, 5919 CrossRef CAS.
- H. Qiu, X. B. Yin, J. Yan, X. Zhao, X. Yang and E. Wang, Electrophoresis, 2005, 26, 687 CrossRef CAS.
- R. S. Martin, A. J. Gawron, S. M. Lunte and C. S. Henry, Anal. Chem., 2000, 72, 3196 CrossRef CAS.
- R. E. Huie and S. Padmaja, Free Radical Res., 1993, 18, 195 CrossRef CAS.
- S. Burney, J. L. Caulfield, J. C. Niles, J. S. Wishnok and S. R. Tannenbaum, Mutat. Res., Fundam. Mol. Mech. Mutagen., 1999, 424, 37 CrossRef CAS.
- R. Radi, J. S. Beckman, K. M. Bush and B. A. Freeman, Arch. Biochem. Biophys., 1991, 288, 481 CrossRef CAS.
- D. Bandyopadhyay, A. Chattopadhyay, G. Ghosh and A. G. Datta, Curr. Med. Chem., 2004, 11, 369 CrossRef CAS.
- R. Radi, J. S. Beckman, K. M. Bush and B. A. Freeman, J. Biol. Chem., 1991, 266, 4244 CAS.
- H. Ischiropoulos and A. B. Al-Medhi, FEBS Lett., 1995, 364, 279 CrossRef CAS.
- M. S. Brown and J. L. Goldstein, Annu. Rev. Biochem., 1983, 52, 223 CrossRef CAS.
- A. Graham, N. Hogg, B. Kalyanaraman, V. O'Leary, V. Darley-Usmar and S. Moncada, FEBS Lett., 1993, 330, 181 CrossRef CAS.
- G. Kojda and D. Harrison, Cardiovasc. Res., 1999, 43, 562 CrossRef CAS.
- I. G. Obrosova, J. G. Mabley, Z. Zsengellér, T. Charniauskaya, O. I. Abatan, J. T. Groves and C. Szabó, FASEB J., 2005, 19, 401 CAS.
- H. Ischiropoulos and J. S. J. Beckman, J. Clin. Invest., 2003, 111, 163 CrossRef CAS.
- T. Keng, C. T. Privalle, G. S. Gilkeson and J. B. Weinberg, Mol. Med., 2000, 6, 779 CAS.
- S. Grinnell, K. Yoshida and H. E. Jasin, Arthritis Rheum., 2005, 52, 80 CrossRef CAS.
- S. Goldstein, J. Lind and G. Merényi, Chem. Rev., 2005, 105, 2457 CrossRef CAS.
- R. Kissner and W. H. Koppenol, J. Am. Chem. Soc., 2002, 124, 234 CrossRef CAS.
- R. Radi, G. Peluffo, M. N. Alvarez, M. Naviliat and A. Cayota, Free Radical Biol. Med., 2001, 30, 463 CrossRef CAS.
- R. Meli, T. Nauser, P. Latal and W. H. Koppenol, J. Biol. Inorg. Chem., 2002, 7, 31 CrossRef CAS.
- C. N. Frankenfeld, M. R. Rosenbaugh, B. A. Fogarty and S. M. Lunte, J. Chromatogr., A, 2006, 1111, 147 CrossRef CAS.
- K. M. Robinson and J. S. Beckman, Methods Enzymol., 2005, 396, 207 CAS.
- M. Abdelgawad and A. R. Wheeler, Adv. Mater., 2007, 19, 133 CrossRef CAS.
- C. L. do Lago, C. A. Neves, D. P. de Jesus, H. D. T. da Silva, J. G. A. Brito-Neto and J. A. Fracassi da Silva, Electrophoresis, 2004, 25, 3825 CrossRef.
- W. K. T. Coltro, E. Piccin, J. A. Fracassi da Silva, C. L. do Lago and E. Carrilho, Lab Chip, 2007, 7, 931 RSC.
- P. Kubáň and P. C. Hauser, Lab Chip, 2005, 5, 407 RSC.
- D. C. Duffy, J. C. McDonald, O. J. A. Schueller and G. M. Whitesides, Anal. Chem., 1998, 70, 4974 CrossRef CAS.
- J. C. McDonald, D. C. Duffy, J. R. Anderson, D. T. Chiu, H. Wu, O. J. A. Schueller and G. M. Whitesides, Electrophoresis, 2000, 21, 27 CrossRef CAS.
- C. L. do Lago, H. D. T. da Silva, C. A. Neves, J. G. A. Brito-Neto and J. A. Fracassi da Silva, Anal. Chem., 2003, 75, 3853 CrossRef.
- W. K. T. Coltro, J. A. Fracassi da Silva and E. Carrilho, Electrophoresis, 2008, 29, 2260 CrossRef CAS.
- M. Pumera, J. Wang, F. Opekar, I. Jelínek, J. Feldman, H. Löwe and S. Hardt, Anal. Chem., 2002, 74, 1968 CrossRef CAS.
- J. Lichtenberg, N. F. de Rooij and E. Verpoorte, Electrophoresis, 2002, 23, 3769 CrossRef CAS.
- J. Tanyanyiwa, E. M. Abad-Villar, M. T. Fernández-Abedul, A. Costa-García, W. Hoffmann, A. E. Guber, D. Herrmann, A. Gerlach, N. Gottschlich and P. C. Hauser, Analyst, 2003, 128, 1019 RSC.
- A. J. Zemann, Electrophoresis, 2003, 24, 2125 CrossRef CAS.
- Y. Marcus, Ion properties, Marcel Dekker, Inc., New York, 1997 Search PubMed.
- D. Y. Boudko, B. Y. Cooper, W. R. Harvey and L. L. Moroz, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2002, 774, 97 CrossRef CAS.
- P. Schmitt-Kopplin, A. V. Garmash, A. V. Kudryavtsev, F. Mezinger, I. V. Perminova, N. Hertkorn, D. Freitag, V. S. Petrosyan and A. Kettrup, Electrophoresis, 2001, 22, 77 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: electropherograms showing the separation of cations at 50 μM (and 25 μM) and anions at 100 μM equimolar concentrations by C4D; electropherogram showing the separation of nitrite and nitrate at 500 μM and 100 μM, respectively, by simultaneous C4D and amperometric detection; effective mobility-scaled electropherograms showing the separation of nitrate and nitrite in a standard mixture and in a peroxynitrite sample. See DOI: 10.1039/b908985c |
| This journal is © The Royal Society of Chemistry 2010 |