High throughput screening of lead utilising disposable screen printed shallow recessed microelectrode arrays
Sebastian J.
Hood
,
Rashid O.
Kadara
,
Dimitrios K.
Kampouris
and
Craig E.
Banks
*
Faculty of Science and Engineering, School of Biology, Chemistry and Health Science, Division of Chemistry and Materials, Manchester Metropolitan University, Chester Street, Manchester, M1 5GD, Lancs, UK. E-mail: c.banks@mmu.ac.uk
First published on 4th November 2009
Abstract
The cathodic stripping voltammetry of lead at disposable screen printed shallow recessed microelectrode arrays has been developed for the first time. The array comprises 6 microdiscs which have radii of 116 (±6) microns which are recessed by 4 microns and are separated by 2500 microns from their nearest neighbour in a hexagonal arrangement. The electroanalytical determination of lead was explored in 0.1 M nitric acid and found that using a 120 s deposition time, a detection limit of 3 μM is feasible which is not possible utilising a screen printed graphite macro-electrode. The sensitivity of this analytical protocol can be tailored by varying the deposition time and it is found that increasing this to 320 s facilitates a limit of detection of 39 nM. This methodology is shown to be feasible for the portable and economical screening of lead in river water samples at the levels indicated by the EC Dangerous Substances Directive (76/464/EEC).
Introduction
Voltammetric methods are becoming the method of choice for the de-centralised screening of target analytes due to portability, short analysis times, low power consumption, favourable cost economics and increased sensitivities and low detection limits.1,2 An application where this methodology is highly favourable is that of trace heavy metal detection.3–9 Amongst toxic heavy metals, lead is one of the most problematic due to its significant health concerns.10 The EC Dangerous Substances Directive (76/464/EEC) recommends that the lead levels in estuarine systems, which rivers and streams flow into, is no more than 0.12 μM; this value varies according to the water hardness.11Mercury coated carbon electrodes were once an electrode of choice for the electroanalytical determination of lead.12,13 Using long deposition times and applying convection, low detection limits (∼10–8 M) are possible. Due to the high toxicity and detrimental impact of mercury on the environment, bismuth modified electrodes were introduced.14–16 The electroanalytical detection of lead which is a key target analyte and new electrode materials are highly desirable which offer simplification, reduction in cost and adequate sensitivities and detection limits for their desired applications. Compton et al. were the first to show the analytical power of cathodic stripping voltammetry utilising boron-doped diamond electrodes with a power ultrasound assisted deposition protocol for the determination of lead in river sediments.17 The application of power ultrasound18 in this context serves to increase mass transport of the target analyte towards the electrode surface. The cathodic stripping of lead is not as sensitive as anodic stripping voltammetry but it is highly selective and thus an increase in the mass transport is required.17 While the work by Compton et al. remains state-of-the-art and is useful for a semi-portable detection strategy, more portable methodologies are demanded. One way of increasing the mass transport is to decrease the size of the electrode from a macroelectrode to a microelectrode or smaller.1,19,20 To further increase the sensitivity, an array of microelectrodes can be readily employed where single microelectrodes are wired in parallel with each electrode diffusionally independent, which generates an amplified signal output allowing a greater sensitivity and lower detection limits.21–23
Recently, we demonstrated proof-of-concept that shallow recessed graphite microelectrode arrays could be fabricated purely by screen printing with reproducible disc radii of 116 (±6) microns.24 This fabrication approach produces economically viable disposable microelectrode arrays which do not require pre-treatment or electrode polishing before use. Using a screen printed 16 microelectrode shallow recessed array, where discs are in a hexagonal pattern separated from their nearest neighbour by 1250 microns, the electroanalytical determination of manganese(II) via cathodic stripping voltammetry was shown to be possible achieving a limit of detection of 81 (±1.2) nM. Changing the arrangement of the array by reducing the number of discs to 6 and increasing the separation to 2500 microns, a detection limit of 64 (±1.5) nM was possible. This low level detection is superior over a range of analytical and electroanalytical methodologies and is due to a reduction in shielding between microdiscs with a greater mass transport possible on the 6 microdisc array.24 In this paper, we report the first analytical use of the 6 microdisc shallow recessed array towards the sensing of lead using cathodic stripping voltammetry.
Experimental section
All chemicals used were of analytical grade and were used as received without any further purification from Sigma-Aldrich. All solutions were prepared with deionised water of resistivity not less than 18.2 MΩ cm−1. Screen printed shallow recessed electrodes were constructed as previously reported.24 These arrays consist of 6 shallow recessed electrodes which are recessed by 4 microns and have a microdisc average radius of 116 (±6) microns arranged in a hexagonal arrangement which are separated from their nearest neighbour by 2500 microns. For comparative purposes, a graphite macroelectrode (3 mm diameter) was employed which is constructed as previously described and has fast electron transfer kinetics.19Voltammetric measurements were carried out using a μ-Autolab III (Eco Chemie, The Netherlands) potentiostat/galvanostat and controlled by Autolab GPES software version 4.9 for Windows XP. All measurements were conducted using a three electrode configuration where the working electrode was the fabricated recessed microelectrode array utilising a carbon-graphite counter electrode and a saturated calomel electrode (SCE) as the reference electrode.
River water was sampled from ‘Jacksons Boat’ bridge over River Mersey, Sale, Cheshire, UK. Analysis of the lead in river water was performed using Graphite Furnace Atomic Absorption Spectroscopy (GFAAS). The GFAAS was a Varian GTA120 Graphite Tube Atomiser and Varian AA240Z Zeeman Atomic Absorption Spectrometer with a lead hollow cathode tube lamp set at 217 nm. The temperature was programmed and was ramped from 85 °C up to 2100 °C. Measurements were performed in triplicate.
Results and discussion
Attention was first turned towards exploring the screen printed shallow recessed microelectrode array towards additions of lead into 0.1 M nitric acid over the range 5 to 70 μM. A conditioning potential at +0.5 V (vs. SCE) for 40 s followed by 120 s deposition at +1.65 V (vs. SCE) was employed in accordance with previous studies17 for comparative purposes. This accumulation step results in the formation of lead dioxide on the graphite electrode surface. Fig. 1 depicts a typical voltammetric profile where a stripping peak is observed at +1.1 V (vs. SCE) in excellent agreement with literature reports.17 The observed stripping peak is due to the electrochemical stripping of lead dioxide back to lead(II):17| 2H2O + Pb2+ ⇌ PbO2 + 4H+ + 2e− |
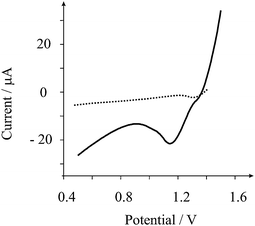 | ||
| Fig. 1 Typical linear sweep cathodic stripping voltammogram of 50 μM lead in 0.1 M nitric acid. Parameters: conditioning at +0.5 V (vs. SCE) for 40 s followed by 120 s deposition at +1.65 V (vs. SCE) employing stripping scan at 25 mV s−1 with a step potential of 5 mV. The arrays consists of 6 shallow recessed electrodes which are recessed by 4 microns and have a microdisc average radius of 116 (±6) microns arranged in a hexagonal arrangement which are separated from their nearest neighbour by 2500 microns. The dotted line is the response of a graphite (3 mm diameter) macroelectrode. | ||
The peak height is taken from the minima of the voltammogram prior to the lead dioxide stripping peak at ∼0.8 V relative to the peak maxima. Analysis of the peak height (IH) from increasing additions of lead is depicted in Fig. 2. A linear response is observed over the range 5 to 30 μM (IH/A = 0.72 A M−1 + 1.87 × 10−5 A; R2 = 0.998; N = 6) beyond which a plateau is observed presumably as the electrode surface becomes saturated. A limit of detection based on 3σ reveals that a detection limit of 3 μM is possible. This linear range and detection limit is competitively similar to that using a 60 s power ultrasound assisted deposition time and employing power ultrasound.17 In comparison, the response of a graphite macroelectrode was sought. This macroelectrode is constructed from the same graphite material19 that is used to fabricate the recessed microelectrode array and was explored at a lead concentration of 50 μM. As shown in Fig. 1, no voltammetric signals are observed indicating that the enhanced mass transport resulting from the geometric structure of the shallow recessed microelectrode array allows the low level determination of lead.
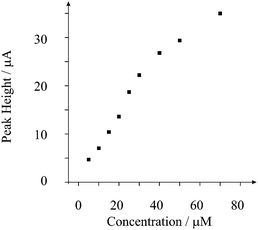 | ||
| Fig. 2 Plot of peak height versus added lead concentration. Experimental parameters as in Fig. 1. | ||
Next, we sought to improve the sensitivity of the screen printed recessed microelectrode array towards the determination of lead by increasing the deposition time. Fig. 3 depicts the response of using a 320 s deposition time. Two linear ranges are observed with the first over the range 0.4 to 1 μM (IH/A = 21.7 A M−1 − 6.2 × 10−6 A; R2 = 0.999; N = 4) and then 1.5 to 10 μM (IH/A = 6.7 A M−1 + 1.0 × 10−5 A; R2 = 0.997; N = 9). Based on the first liner response, a limit of detection (3σ) was found to correspond to 3.9 × 10−8 M. This detection limit is competitive to previous reports of lead sensing using amide-cyclam-functionalised silica-modified carbon paste electrodes,25 nanoparticle modified boron-doped diamond electrodes26 and nanomaterial/ionophore-based electrodes.27 In comparison to current state-of-the-art,17 this work is highly comparable where detection limits of the order 10−8 M were possible using a 240 s deposition time with intense power ultrasound. Our method has a slightly longer deposition time (320 s) but does not require convection in the form of power ultrasound and is greatly simplified. This analytical protocol based on the cathodic stripping voltammetry of lead is useful for real analytical challenges, since it is extremely selective with chromium, nickel, cadmium and zinc having no measurable effect upon the technique's response to lead. In the case of copper and iron, these may be detected via cathodic stripping voltammetry but their stripping peaks occur at well resolved potentials from that of lead.17 Due to this high elemental selectivity, this voltammetric method utilising sono-electrochemistry in conjugation with boron-doped diamond electrodes has been shown to be successful for the detection of lead in river sediments;17 consequently, we now turn to exploring the feasibility of using this novel electrochemical sensing platform for the screening of lead in river water.
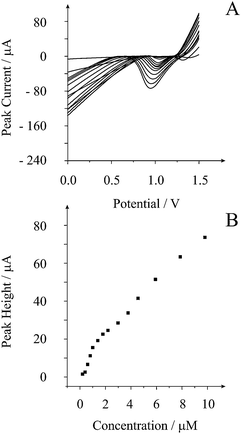 | ||
| Fig. 3 Typical linear sweep cathodic stripping voltammogram (A) from additions of lead in 0.1 M nitric acid with the analysis of peak height vs. added lead concentration (B). Parameters: conditioning at +0.5 V (vs. SCE) for 40 s followed by 340 s deposition at +1.65 V (vs. SCE) employing stripping scan at 25 mV s−1 with a step potential of 5 mV. | ||
River water was sampled from ‘Jacksons Boat’ bridge over River Mersey, Sale (Cheshire, UK). The sample's pH was changed to pH 1.3 with a few drops of concentrated nitric acid and did not undergo any further treatment. The parameters as used in Fig. 3 were employed with a standard addition protocol adopted. Based on this measurement, the amount of lead in the river water sample was found to correspond to 0.1 (±0.04) μM. The analysis of lead in the river sample was repeated and analysed via Graphite Furnace Atomic Absorption Spectroscopy (GFAAS) which determined a value of 0.24 (±0.02) μM; this level is in agreement with that reported in ref. 28 and that of the EC Dangerous Substances Directive (76/464/EEC).11 It is important to note that the GFAAS represents the total amount of lead in the river sample while the voltammetric method will measure the free lead concentration. In the sample, the lead may be absorbed in microparticle sediment deposits by the presence of micro-organisms or natural dissolved organic matter.29
In reality, the high throughput screening of lead at the river side is desirable. We envisage that at the site of sampling, using the screen printed recessed microelectrode array and the parameters used in Fig. 3, if voltammetric peaks are observed, indicating that low micro-molar levels of lead are present in the sample, the sample should be transported from the river side for further detailed analysis. Analysis of ten repetitive measurements of a river sample spiked with 0.4 μM lead was found to produce a percentage relative standard deviation of 8%, suggesting this method’s applicability. Future work in this area will be conducted which requires a substantial number of real water samples to be analysed.
Conclusions
The determination of lead has been shown to be feasible at disposable screen printed shallow recessed microelectrode arrays. Through utilising a 120 s deposition time, a detection limit of 3 μM is feasible. At this low lead level, no electroanalytical signal is observable on a graphite macroelectrode, indicating the increased mass transport on the recessed microelectrode array. The detection limit and range may be lowered by increasing the deposition time to 320 s allowing a detection limit of 39 nM. The increased mass transport at the microelectrode array precludes the need for convection to be used during the deposition step which is required using other electrodes. Additionally, electrode pre-treatments such as polishing is avoided due to the disposable nature of the electrode. This analytical protocol was explored and shown to be feasible for the sensing of lead in river water samples at the levels indicated by the EC Dangerous Substances Directive (76/464/EEC). Future work will involve the extension of this work towards the sensing of lead in drinking water samples.References
- R. G. Compton; C. E. Banks, Understanding Voltammetry; World Scientific Ltd, 2007 Search PubMed.
- J. Wang, Analytical Electrochemistry, Wiley Blackwell, 2006 Search PubMed.
- D. W. M. Arrigan, Analyst, 1994, 119, 1953 RSC.
- C. E. Banks, J. Kruusma, M. E. Hyde, A. Salimi and R. G. Compton, Anal. Bioanal. Chem., 2004, 379, 277 CrossRef CAS.
- V. Beni, V. I. Ogurtsov, N. V. Bakunin, D. W. M. Arrigan and M. Hill, Anal. Chim. Acta, 2005, 552, 190 CrossRef CAS.
- D. Omanovic, Z. Kwokal, A. Goodwin, A. Lawrence, C. E. Banks and S. Komorsky-Lovric, J. Iranian Chem. Soc., 2006, 3, 128 Search PubMed.
- K. E. Toghill, G. G. Wildgooses, A. Moshar, C. Batchelor-McAuley and R. G. Compton, Electroanalysis, 2008, 20, 1731 CrossRef CAS.
- V. Cuculic and M. Branica, Analyst, 1996, 121, 1127 RSC.
- I. Svancara and K. Vytras, Chem. Listy, 2006, 100, 90 CAS.
- W. Yantasee, Y. Lin, K. Hongsirikarn, G. E. Fryxell, R. Addleman and C. Timchalk, Environ. Health Perspect., 2007, 115, 1683 CrossRef CAS.
- http://www.environment-agency.gov.uk/static/documents/EQS_tables.pdf; accessed August 2009.
- J. Wang and B. M. Tian, Anal. Chem., 1992, 64, 1706 CrossRef CAS.
- M. Wojciechowski and J. Balcerzak, Anal. Chem., 1990, 62, 1325 CrossRef CAS.
- J. Wang, J. M. Lu, S. B. Hocevar and B. Ogorevc, Electroanalysis, 2001, 13, 13 CrossRef CAS.
- J. Wang, Electroanalysis, 2005, 17, 1341 CrossRef CAS.
- J. Kruusma, C. E. Banks and R. G. Compton, Anal. Bioanal. Chem., 2004, 379, 700 CrossRef CAS.
- A. J. Saterlay, C. Agra-Gutierrez, M. P. Taylor, F. Marken and R. G. Compton, Electroanalysis, 1999, 11, 1083 CrossRef CAS.
- C. E. Banks and R. G. Compton, ChemPhysChem, 2003, 4, 169 CrossRef CAS.
- R. O. Kadara, N. Jenkinson and C. E. Banks, Electrochem. Commun., 2009, 11, 1377 CrossRef CAS.
- T. J. Davies, C. E. Banks and R. G. Compton, J. Solid State Electrochem., 2005, 9, 797 CrossRef CAS.
- S. J. Hood, D. K. Kampouris, R. O. Kadara, N. Jenkinson, F. Javier del Campo, F. X. Munoz and C. E. Banks, Analyst, 2009, 134, 2301 RSC.
- C. Batchelor-McAuley, C. E. Banks, A. O. Simm, T. G. J. Jones and R. G. Compton, Analyst, 2006, 131, 106 RSC.
- S. Fletcher and M. D. Horne, Electrochem. Commun., 1999, 1, 502 CrossRef CAS.
- R. O. Kadara, N. Jenkinson and C. E. Banks, Sens. Act. B., 2009, 11, 1377 CAS.
- S. Goubert-Renaudin, M. Moreau, C. Despas, M. Meyer, F. Denat, B. Lebeau and A. Walcarius, Electroanalysis, 2009, 21, 1731 CrossRef CAS.
- K. E. Toghill, L. Xiao, G. G. Wildgoose and R. G. Compton, Electroanalysis, 2009, 21, 1113 CrossRef CAS.
- D. W. Pan, Y. E. Wang, Z. P. Chen, T. T. Lou and W. Qin, Anal. Chem., 2009, 81, 5088 CrossRef CAS.
- http://www.water.org.uk/static/files_archive/1Lead_-_Water_UK.pdf; accessed August 2009.
- G. E. Millward and Y. P. Liu, Sci. Total Environ., 2003, 313–316, 613 CrossRef.
| This journal is © The Royal Society of Chemistry 2010 |