Penetrative DNA intercalation and G-base selectivity of an organometallic tetrahydroanthracene RuII anticancer complex†
Hong-Ke
Liu
*a,
John A.
Parkinson
c,
Juraj
Bella
e,
Fuyi
Wang
d and
Peter J.
Sadler
*b
aJiangsu Key Laboratory of Biofunctional Materials, School of Chemistry, Nanjing Normal University, Nanjing, China. E-mail: liuhongke@njnu.edu.cn; Tel: +86-2585891651
bDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, UK CV4 7AL. E-mail: P.J.Sadler@warwick.ac.uk; Fax: +44-2476523819; Tel: +44-2476523818
cWestCHEM Department of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, UK G1 1XL
dBeijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, China
eSchool of Chemistry, University of Edinburgh, King's Buildings, West Mains Road, Edinburgh, UK EH9 3JJ
First published on 11th June 2010
Abstract
The organometallic RuII arene complex [(η6-tha)Ru(en)Cl]+ (1), where tha = tetrahydroanthracene and en = ethylenediamine, is potently cytotoxic towards cancer cells. We have used a combination of HPLC, ESI-MS, 1D- and 2D-NMR, including [1H, 1H] ROESY, NOESY, [1H, 15N] HSQC (using 15N-1), and [1H, 31P] experiments to elucidate the role of the non-aromatic, bulky rings of tha in adducts with the DNA hexamer d(CGGCCG), since DNA is a potential target for this drug. Reactions of 1 with single-stranded d(CGGCCG) gave rise to ruthenation at each of the three G bases, whereas reactions of the duplex d(CGGCCG)2 with 1 mol equiv. 1 led to exclusive ruthenation of G3 and G6 (and G9, G12) and not G2 (or G8). Addition of a second mol equiv. of 1 gave di-ruthenated adducts (major sites G3/G6, G6/G9, G2/G6), and on reaction with a third mol equiv. tri-ruthenation (G2, G3/G6/G12).The NMR data are indicative of the coordinative binding of Ru-tha specifically to G3 and G6, together with penetrative intercalation of the bulky non-coordinated tha rings B and C of 1′, selectively between two base pairs G3/C10:C4/G9 and G6/C7:C5/G8. Intercalation at GpC base steps by tha has a lower energy penalty compared to intercalation at GpG base steps, thereby allowing accommodation of tha. Mono-intercalation of tha reduced the strength of H-bonding between en-NH and GO6. These differences in structural distortions compared to cisplatin induced by the coordinative binding of Ru-tha to GN7 may contribute to the differences in mechanism of action, including protein recognition of the metallated lesions, and lack of cross resistance.
Introduction
There is currently much interest in the design and mechanism of action of ruthenium anticancer complexes.1–7 Structural distortions of DNA are thought to play a major role in the anticancer activity of many ruthenium and other transition metal complexes.1,8–23 It is apparent that distortions induced in DNA by [(η6-arene)Ru(en)Cl]+ organometallic ruthenium(II) arene anticancer complexes1,21,24–28 (en = ethylenediamine) differ significantly from those induced by cisplatin.9,10 In particular the arene appears to play a significant role in DNA interactions and the cytotoxicity of complexes shows a strong dependence on the arene.19,22,23,29,31 The cytotoxic activity of these ruthenium arene complexes appears to increase with the size of the coordinated arene:1,24,25p-cymene (Ru-cym) < biphenyl (Ru-bip) < dihydroanthracene (Ru-dha) < tetrahydroanthracene (Ru-tha, 1, Fig. 1), the latter complex being as cytotoxic as the clinical platinum drug cisplatin. If the arene is extended, the possibility arises of intercalation between DNA base-pairs. Studies of the interactions of ruthenium complexes Ru-cym and Ru-bip with the 6-mer duplex d(CGGCCG)2 revealed that the binding of Ru to GN7 is accompanied by strong H-bonding between GO6 and en-NH, and that the arene ligand distorts the duplex either via steric interactions (Ru-cym) or via intercalation19,22,23,32,38 (Ru-bip), which may explain why the IC50 values of complexes Ru-cym and Ru-bip are similar.1,24,25 Further work23 revealed a unique mode of binding of Ru-bip to a 14-mer DNA duplex: the monofunctional fragment {(η6-biphenyl)Ru(en)}2+ is highly specific for GN7, although mobile at elevated temperature (migrating to other G residues). The uncoordinated phenyl ring of bip can be involved in π–π stacking with DNA bases either via classical intercalation19,35–38 between the bases, or with a partially extruded T base.23,39,40![Structures and NMR numbering schemes for [(η6-tha)Ru(en)Cl]+ (1) and for the 5′-d(CpGp) fragment of the hexamer d(CGGCCG); single-strand (ss) I and duplex II.](/image/article/2010/SC/c0sc00175a/c0sc00175a-f1.gif) | ||
| Fig. 1 Structures and NMR numbering schemes for [(η6-tha)Ru(en)Cl]+ (1) and for the 5′-d(CpGp) fragment of the hexamer d(CGGCCG); single-strand (ss) I and duplex II. | ||
Arene–base stacking may play a role in determining the rates of reactions of RuII arene complexes with DNA, as appears to be the case for mononucleotides. We have shown previously that the tha and bip arenes can exert slightly different effects on the chemical reactivity of these RuII complexes and on distortions induced in DNA.26,29,30 For example the rate of reaction of Ru-bip with cGMP is ca. 2× slower than that of 1,27 but Ru-bip induces similar unwinding of DNA as complex 1 (14°).26 Both of these complexes can potentially intercalate into DNA, leading to “downstream” effects on DNA processing and repair mechanisms. Ru-bip is an aromatic intercalator,22,23,41 in which all the protons of the extended ring are within the aromatic plane, and can form π–π interactions with bases; however, the extended rings B and C of tha in 1 are bulky non-aromatic groups; the three rings A, B and C are not in the same plane and the H5,8 and H9,10 protons are located above or below the arene ring plane by nearly 0.9 Å. The extended rings B and C cannot form π–π interactions with bases in the same way that aromatic intercalators do and so Ru-tha is more sterically demanding than Ru-bip. It is reasonable to predict that the intercalative interactions between duplex DNA and Ru-tha or Ru-bip should be quite different. The Ru-tha complex 1 is 10 times more toxic to cancer cells than Ru-bip.24,25
Bulky substituents at the sites of DNA lesions may activate nucleotide-excision repair;42,43 however, reports of intercalation by bulky molecules are rare. Gomez-Pinto et al.44 have shown that intercalation of a modified nucleotide containing a cholesterol derivative into a DNA decamer induces DNA distortions which are different from those induced by aromatic intercalators.45,46
Threading intercalation has attracted recent attention because the intercalator occupies and interacts strongly with both the minor and major grooves of DNA simultaneously. This has been observed for polyaromatic intercalators,47–49 dinuclear metallointercalators,50–53 and platinum complexes with aromatic side-arms, such as acridine-9-ylthiourea.54–56 As a result, threading intercalative interactions promise high DNA binding affinity and specificity, a slow rate of dissociation, and an enhanced ability to block DNA–protein interactions.50,57
It is important to understand the mode of interaction between [(η6-tha)Ru(en)Cl]+ (1) and DNA since a major contributor to its high potency appears to be the lack of repair of the lesions formed on DNA by this complex,29i.e. lack of recognition by repair enzymes. In the present work we have investigated the role of the extended non-aromatic bulky tha in interactions of [(η6-tha)Ru(en)Cl]+ (1) with the single-stranded hexamer d(CGGCCG) and double-stranded duplex d(CGGCCG)2 using a wide variety of experimental techniques including HPLC, ESI-MS and 1D 1H and 2D [1H,1H] ROESY, NOESY, [1H, 15N] HSQC (using 15N-1), and [1H, 31P] NMR spectroscopy.
Results
Scheme 1 indicates the reaction pathways that were followed during the course of this study. HPLC enabled separation of single DNA strands even for reactions of the duplex. The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2:1 and 3:1 1/I mixtures, 1.1:1 and 2:1 1/II mixtures were studied by HPLC and ESI-MS, and 1.1:1, 2:1 and 3:1 1/II mixtures were studied by 1D 1H and 2D [1H, 1H] TOCSY NMR experiments. The 1.1:1 1/II mixture was studied by 1D 1H, 2D 15N-decoupled [1H, 1H] ROESY, NOESY, 15N-edited [1H, 1H] TOCSY and NOESY, 2D 15N-decoupled [1H, 15N] and [1H, 31P] HSQC NMR experiments using 15N-en labelled 1. A near-complete NMR spectral assignment of the NOESY NMR spectrum of the 1.1:1 1/II mixture was achieved, although its complexity precluded full structure determination. Assignments were made possible by the known selectivity of 1 for guanines, and the localization of structural perturbations to residues close to the ruthenated G residue. Thus, sequential assignments along each strand always led to cross-peaks largely identical to those of the non-ruthenated duplex. Despite extensive overlap of NOE cross-peaks, little ambiguity in the assignments of individual resonances was found, with cross-validation of signal assignments from related connectivities being possible, with the help of [1H, 31P] HSQC, [1H, 15N] HSQC and 15N-edited [1H, 1H] NOESY experiments and HPLC-MS data. 2D 15N-decoupled [1H, 1H] ROESY and NOESY and 15N-decoupled [1H, 15N] HSQC NMR data were also acquired for 2:1 and 3:1 1/II mixtures, but the spectra were too complex for interpretation.
:1, 2:1 and 3:1 1/I mixtures, 1.1:1 and 2:1 1/II mixtures were studied by HPLC and ESI-MS, and 1.1:1, 2:1 and 3:1 1/II mixtures were studied by 1D 1H and 2D [1H, 1H] TOCSY NMR experiments. The 1.1:1 1/II mixture was studied by 1D 1H, 2D 15N-decoupled [1H, 1H] ROESY, NOESY, 15N-edited [1H, 1H] TOCSY and NOESY, 2D 15N-decoupled [1H, 15N] and [1H, 31P] HSQC NMR experiments using 15N-en labelled 1. A near-complete NMR spectral assignment of the NOESY NMR spectrum of the 1.1:1 1/II mixture was achieved, although its complexity precluded full structure determination. Assignments were made possible by the known selectivity of 1 for guanines, and the localization of structural perturbations to residues close to the ruthenated G residue. Thus, sequential assignments along each strand always led to cross-peaks largely identical to those of the non-ruthenated duplex. Despite extensive overlap of NOE cross-peaks, little ambiguity in the assignments of individual resonances was found, with cross-validation of signal assignments from related connectivities being possible, with the help of [1H, 31P] HSQC, [1H, 15N] HSQC and 15N-edited [1H, 1H] NOESY experiments and HPLC-MS data. 2D 15N-decoupled [1H, 1H] ROESY and NOESY and 15N-decoupled [1H, 15N] HSQC NMR data were also acquired for 2:1 and 3:1 1/II mixtures, but the spectra were too complex for interpretation.
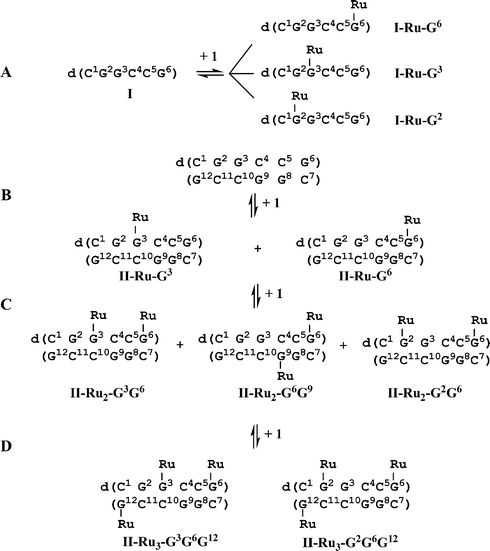 | ||
| Scheme 1 (A) Reaction of single-stranded (ss) hexamer I (0.1 mM) with 1 mol equiv. of 1 in H2O, 310 K for 48 h, gives three mono-ruthenated I (I-Ru-G2, I-Ru-G3, I-Ru-G6). (B) Reaction of double-stranded (ds) hexamer II (0.3 mM, 0.1 M NaClO4) with 1.1 mol equiv. of 1 in 90% H2O/10% D2O gives rise to two mono-ruthenated duplexes II-Ru-G3 and II-Ru-G6 as products. Addition of a second mol equiv. of 1 results in di-ruthenated duplexes, including II-Ru2-G3G6, II-Ru2-G6G9 and II-Ru2-G2G6 as main products. Addition of a third mol equiv. of 1 results in two tri-ruthenated duplexes II-Ru3-G3G6G12 and II-Ru3-G2G6G12 as main products. Ru = {(η6-tha)Ru(en)}2+, 1′. For structure of 1, see Fig. 1. | ||
HPLC and ESI-MS characterization of products from ss-DNA I + 1
A 100 μM aqueous solution of 1 was incubated with I at 310 K at Ru:I molar ratios of 1:1, 2:1 and 3:1 for 48 h in the dark, and these were then analyzed by HPLC. The low ionic strength (5.1 × 10−4 M) ensures that this self-complementary oligonucleotide remains largely single-stranded (calculated melting temperature 264 K) under these conditions.58 New peaks were observed for each reaction (Fig. 2 and Table S1†), and the adducts associated with them were identified subsequently by ESI-MS. The peaks for the observed negative ions are listed in Table S1.† Reaction at a Ru:I molar ratio of 1:1 resulted in three mono-ruthenated products together with three di-ruthenated products. Reaction at a Ru:I molar ratio of 2:1 resulted in three di-ruthenated products together with a tri-ruthenated product. Reaction at a Ru:I molar ratio of 3:1, gave only one main HPLC peak corresponding to a tri-ruthenated product.
![HPLC chromatograms for reaction of [(η6-tha)Ru(en)Cl]+ (1) with single-stranded (ss) d(CGGCCG) (I) (0.1 mM in H2O) at Ru : I mol ratios of (a) 1 : 1, (b) 2 : 1, and (c) 3 : 1, and for reaction of 1 with duplex d(CGGCCG)2 (II) (0.34 mM, 0.1 M NaClO4, 90% H2O/10% D2O) at a Ru : II mol ratio of (d) 1.1 : 1 and (e) 2 : 1. The mono-ruthenated duplex II-Ru1 elutes as mono-ruthenated ss-DNA I (I-Ru-G3 and I-Ru-G6; species I-Ru-G3 and I-Ru-G9 are identical, as are I-Ru-G6 and I-Ru-G12); di-ruthenated duplex II-Ru2 elutes as mono-ruthenated ss-DNA I (I-Ru-G3 and I-Ru-G6, see (d)), and di-ruthenated ss-DNA I (I-Ru2, see (e)). It is notable that G2 is readily ruthenated for single strand I (see I-Ru-G2 in (a)) but not for duplex II in (d). Little ruthenation on G8 was observed when 1 mol equiv. of 1 was added to mono-ruthenated duplexes II-Ru-G3 and II-Ru-G6 (e). Ru = {(η6-tha)Ru(en)}2+ (1′), and is bound to G3N7 or G6N7; for DNA sequence, see Fig. 1 and Scheme 1.](/image/article/2010/SC/c0sc00175a/c0sc00175a-f2.gif) | ||
| Fig. 2 HPLC chromatograms for reaction of [(η6-tha)Ru(en)Cl]+ (1) with single-stranded (ss) d(CGGCCG) (I) (0.1 mM in H2O) at Ru:I mol ratios of (a) 1:1, (b) 2:1, and (c) 3:1, and for reaction of 1 with duplex d(CGGCCG)2 (II) (0.34 mM, 0.1 M NaClO4, 90% H2O/10% D2O) at a Ru:II mol ratio of (d) 1.1:1 and (e) 2:1. The mono-ruthenated duplex II-Ru1 elutes as mono-ruthenated ss-DNA I (I-Ru-G3 and I-Ru-G6; species I-Ru-G3 and I-Ru-G9 are identical, as are I-Ru-G6 and I-Ru-G12); di-ruthenated duplex II-Ru2 elutes as mono-ruthenated ss-DNA I (I-Ru-G3 and I-Ru-G6, see (d)), and di-ruthenated ss-DNA I (I-Ru2, see (e)). It is notable that G2 is readily ruthenated for single strand I (see I-Ru-G2 in (a)) but not for duplex II in (d). Little ruthenation on G8 was observed when 1 mol equiv. of 1 was added to mono-ruthenated duplexes II-Ru-G3 and II-Ru-G6 (e). Ru = {(η6-tha)Ru(en)}2+ (1′), and is bound to G3N7 or G6N7; for DNA sequence, see Fig. 1 and Scheme 1. | ||
HPLC and ESI-MS characterization of products from reaction of duplex II + 1
An aqueous solution of 1 was incubated with duplex II in 0.1 M NaClO4 at ambient temperature for 24 h at a Ru:II molar ratio of 1.1:1 in an NMR tube in the dark. This gave rise to HPLC peaks which were identified by ESI-MS as ss-DNA I and two mono-ruthenated single-stranded products (see Fig. 2(d), Table S1†), with relative peak area ratios of 2:1. Another equimolar amount of 1 was then added to give a Ru:II molar ratio of 2:1, and was kept at ambient temperature in the dark for 48 h. This gave HPLC peaks which were identified by ESI-MS as two mono-ruthenated and two di-ruthenated single-stranded products (see Fig. 2(e) and Table S1†).
NMR characterization of products
Assignments of the 1H NMR peaks for the ruthenated DNA duplexes were made on the basis of established methods developed for studying right-handed B-DNA duplexes by NMR spectroscopy.59 The assignments of the 1H NMR resonances of free DNA duplex II have been reported by Lam and Au-Yeung60 and the 1H and 31P chemical shifts are listed in Table S2.† Terminal (3′) base resonance assignments were identified from NOESY NMR data sets and were based on ordering of the H2′ and H2′′ proton chemical shifts (H2′ > H2′′) compared with the other nucleotide units (H2′ < H2′′). The resonance assignments of backbone 31P and sugar ring 1H (H4′ and H3′) resonances for free and mono-ruthenated DNA duplexes were achieved by [1H, 31P] HSQC NMR experiments61 and the H4′n and H3′n−1 protons were assigned by correlation to their respective 31Pn resonances. The assignments of ruthenated G*H8 and 1′-en-NH21H (NHu and NHd, see Fig. 1 for labelling) resonances of mono-ruthenated DNA duplexes were achieved by reference to 2D [1H, 15N] HSQC and 15N-edited [1H, 1H] NOESY NMR data. The NMR chemical shifts of the 1H and 31P resonances associated with these two mono-ruthenated duplex adducts are listed in Table S3 (II-Ru-G3), Table S4 (II-Ru-G6) and Table S2 (free II).† The assignments of H1,4, H2,3, H9,10, H5,8, H6,7 and 1′-en-NH2 (NHu and NHd, see Fig. 1 for labelling) 1H NMR resonances of {(η6-tha)Ru(en)}2+ (1′) were achieved by 2D [1H, 15N] HSQC, 15N-edited [1H, 1H] NOESY and [1H, 1H] NOESY experiments27,28 and are listed in Table 1 and Fig. 5. The assignments of H2,3 and H1,4 NMR resonances of 1′ were achieved by correlations to the 1′-en-NHu resonances in [1H, 1H] NOESY NMR data, where the H9,10 protons were assigned by correlation to the H1,4 resonances, and H5,8 protons by correlation to the H9,10 and H6,7 resonances.27,28:1 1/II reaction product 1′-II, in Ru-tha-9EtG (1′-9EtG)a,b and Ru-tha-5′GMP (1′-GMP) adductsa,b
| Complex | δ(1H) (Δδ) | |||||||
|---|---|---|---|---|---|---|---|---|
| en-CH2 | NHd | H1,4 | H2,3 | NHu | H5,8 | H9,10 | H6,7 | |
| a For atom labels, see Fig. 1 and Scheme 1. b Ref. 28. c Δδ = δ(1′-II) − δ(1) (≥0.04 ppm). d This assignment is based on a NOESY experiment. e At 283 K. f At 339 K. g At 318 K. h Δδ = δ(1′-9EtG) − δ(1) (≥0.04 ppm). i Δδ = δ(1′-GMP) − δ(1) (≥0.04 ppm). | ||||||||
| 1 | 2.23/2.43 | 3.60/3.71 | 5.50 | 5.61 | 6.19/6.29 | 2.62 | 3.18 | 5.75 |
| 1′ -II | 2.34/2.45 (0.11/)c | 3.86/3.95d (0.26/0.24)c | 6.04 (0.54)c | 5.97 (0.36)c | 6.47/6.56 (0.18/0.27)c | 2.74 (0.12)c | 4.18 (1.00)c | 5.84 (0.09) c |
| 1′-9EtG b,f | 2.07/2.40 (−0.16)h | na | 5.85 (0.35)h | 6.24 (0.63)h | na | 2.55 (−0.07)h | 3.20 | 5.66 (−0.09)h |
| 1′-GMP b,g | 2.10/2.40 (−0.13)i | na | 6.16 (0.66)i | 6.16 (0.55)i | na | 3.12 (0.50)i | 4.01 (0.83)i | 5.65 (−0.10)i |
NMR of 1.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1, 2:1 and 3:1 1/II reactions
:1, 2:1 and 3:1 1/II reactions
Fig. S1 shows the imino and aromatic region of the 800 MHz 1D 1H NMR spectrum of DNA duplex II in the absence (Fig. S1A†) and presence of 1 mol (Fig. S1B†), 2 mol (Fig. S1C†) and 3 mol (Fig. S1D†) equiv. of 1. Reaction with 1.1 mol equiv. of 1 resulted in the formation of a number of new peaks for II (especially near 8.5 ppm (G*H8), 13.0–13.6 ppm (imino) and 6.3–6.7 ppm (NHu-1′, 1′ is the bound complex 1, {(η6-tha)Ru(en)}2+), Fig. S1B†). Two imino 1H NMR resonances were shifted to low-field by +0.04 ppm (G3*, mono-ruthenated G3 base) and +0.14 ppm (G9), and two imino 1H resonances were shifted to high-field by −0.07 ppm (G6*, mono-ruthenated G6 base) and −0.04 ppm (G12), relative to the free duplex II (Figs. S1 and S2, Tables S3, S4 and S2†). Reaction of the second mol equiv. of 1 with the mono-ruthenated duplexes resulted in a notable increase in intensities of the new peaks, especially G*H8 (near 8.5 ppm), NHu-1′ (6.3–6.7 ppm), H5 and H1′ resonances; the intensities of imino and H8, H6 resonances of free II all decreased (Fig. S1C†). Reaction of the third mol equiv. of 1 with the di-ruthenated duplexes resulted in an increase in the intensities of new peaks, e.g. at 8.8 ppm (Fig. S1D†). The resonances of CH5, CH6, H1′ and G*H8 moved to low field by up to +0.3 ppm, and the peaks for imino protons almost disappeared.
2D [1H, 1H] TOCSY NMR of 1.1:1, 2:1 and 3:1 1/II reactions
The 2D TOCSY NMR spectrum of the 1.1:1 1/II reaction mixture clearly showed the existence of cross-peaks for the two mono-ruthenated duplexes, as seen for example in the aromatic region in Fig. 3B. Two sets of H5-H6 cross-peaks were detected for C4, C5, C7 and C10 residues. The proportions of Ru-IIa and Ru-IIb at 283 K were determined by integration of the TOCSY cross-peak volumes of C4-H5/C4-H6 of Ru-IIa, and C5-H5/C5-H6 of Ru-IIb, and the HPLC peak areas for I-Ru-G3 and I-Ru-G6 (see Fig. 2d). This gave a Ru-IIa:Ru-IIb ratio of 2:1 (±10%). Other species account for less than 10% of the total DNA. The 2D TOCSY NMR spectrum of the 2:1 1/II reaction mixture shows that peaks for other new species are present but not all can be assigned due to the complexity of the spectrum (Fig. 3C). It was notable that the intensities of the CH5-CH6 cross-peaks for C5/C11 residues of free II decreased remarkably, but those of the CH5-CH6 cross-peaks for C1′, C4′ and C5′ residues of the ruthenated species increased markedly. The 2D TOCSY NMR spectrum of the 3:1 1/II reaction mixture shows that the CH5-CH6 cross-peaks for C5/C11 residues of free II almost completely disappeared (Fig. 3D).
![2D [1H, 1H] TOCSY NMR spectrum in the cytosine H5/H6 cross-peak region for free duplex II (A), 1.1 : 1 1/II mixture (B), 2 : 1 1/II mixture (C) and 3 : 1 1/II mixture (D) of ruthenium complex 1 and duplex II (0.34 mM, 0.1 M NaClO4) in 90% H2O/10% D2O at 283 K. Note that 2 sets of resonances are observed for the C4, C5, C7 and C10 residues, suggesting the presence of two mono-ruthenated products in the 1.1 : 1 1/II mixture (B). Significant changes are observed for H5/H6 resonances of C5 and C11 bases when the Ru:II ratio is increased from 1.1 : 1 to 3 : 1, and the H5/H6 cross-peaks of C5 and C11 bases disappear when the Ru : II ratio reaches 3 : 1, suggesting that all the G6 and G12 bases are ruthenated in the 1/II mixture 3 : 1. Assignment: C1′, C1-H5/H6 cross-peak of ruthenated species; C4′, C4-H5/H6 cross-peak of ruthenated species; C5′, C5–H5/H6 cross-peak of ruthenated species. Assignments are based on the 2D [1H, 1H] NOESY NMR spectrum (Tables S2–S4) and HPLC results (Fig. 2); for DNA sequence, see Scheme 1.](/image/article/2010/SC/c0sc00175a/c0sc00175a-f3.gif) | ||
| Fig. 3 2D [1H, 1H] TOCSY NMR spectrum in the cytosine H5/H6 cross-peak region for free duplex II (A), 1.1:1 1/II mixture (B), 2:1 1/II mixture (C) and 3:1 1/II mixture (D) of ruthenium complex 1 and duplex II (0.34 mM, 0.1 M NaClO4) in 90% H2O/10% D2O at 283 K. Note that 2 sets of resonances are observed for the C4, C5, C7 and C10 residues, suggesting the presence of two mono-ruthenated products in the 1.1:1 1/II mixture (B). Significant changes are observed for H5/H6 resonances of C5 and C11 bases when the Ru:II ratio is increased from 1.1:1 to 3:1, and the H5/H6 cross-peaks of C5 and C11 bases disappear when the Ru:II ratio reaches 3:1, suggesting that all the G6 and G12 bases are ruthenated in the 1/II mixture 3:1. Assignment: C1′, C1-H5/H6 cross-peak of ruthenated species; C4′, C4-H5/H6 cross-peak of ruthenated species; C5′, C5–H5/H6 cross-peak of ruthenated species. Assignments are based on the 2D [1H, 1H] NOESY NMR spectrum (Tables S2–S4†) and HPLC results (Fig. 2); for DNA sequence, see Scheme 1. | ||
2D [1H, 15N] HSQC NMR of the 1.1:1 1/II reaction
These experiments allowed detection of NMR peaks specifically for the {(η6-tha)Ru(15N-en)}2+ fragment. These are commonly difficult to resolve in normal 1H NMR experiments. One major new species was detected by 2D [1H, 15N] HSQC NMR analysis of the 1.1:1 mixture of duplex II and 15N-1 (the 15N-en labelled complex 1) at 283 K in 90% H2O/10% D2O (Fig. S3†). Peaks were assignable to en-NHu resonances (the NH protons oriented towards the coordinated arene ring, see Fig. 1 and Table S5†) of mono-ruthenated duplexes Ru-IIa and Ru-IIb (Ru = {(η6-tha)Ru(en)}2+ (1′)). No cross-peaks for en-NHu resonances of Ru-IIa and Ru-IIb were detectable after the equilibrium mixture had been freeze-dried and re-dissolved in D2O at 283 K. The en-NHd resonances of both Ru-IIa and Ru-IIb were not observed in either H2O or D2O solutions. In contrast, the en-NHu and en-NHd resonances of unreacted 1 were detected in 90% H2O (Fig. S3†). The assignments are listed in Table S5.†
2D [1H, 31P] HSQC NMR of 1.1:1 1/II reaction
The backbone phosphate 31P (−0.6 to −1.4 ppm) to sugar ring H3′ (5.3–4.6 ppm) and H4′ (4.6–4.0 ppm) HSQC connectivities for free duplex II and the 1.1:1 1/II reaction are shown in Fig. 4 and the assignments are listed in Tables S3 and S4.† Compared to free duplex II, the 31P/H4′ cross-peaks for C4 (peak e) and G6* (peak i) residues, 31Pn+1/H3′n cross-peaks for G2-G3* (peak d), G3*-C4 (peak f) and C4-C5 (peak h) residues were shifted to give new peaks, but 31P/H4′ cross-peaks for G3*, C4 and C5 (peak g) and 31Pn+1/H3′n cross-peaks for C5-G6* (peak j) residues were too broad to assign. Decreased intensities of 31P/H4′ and 31Pn+1/H3′n cross-peaks were found for G2/G8 (peak b), G3/G9 (peak c), and C4/C10 (peak d) residues. These results are consistent with the HPLC-MS and 2D TOCSY NMR data.
![2D [1H, 31P] HSQC NMR spectra of (A) duplex II and (B) 1.1 : 1 1/II mixture (0.34 mM, 0.1 M NaClO4 at 283 K, pH 7.0) in 90% H2O/10% D2O, showing the backbone 31P (−1.4 to −0.60 ppm) to sugar ring H3′ (5.2–4.6 ppm) and H4′ (4.6–4.0 ppm) connectivities. The circles indicate Cn-31P/H4′, Cn-31P/Cn−1-H3′ and Cn-31P/Gn−1-H3′ or Gn-31P/H4′, Gn-31P/Gn−1-H3′ and Gn-31P/Cn−1-H3′ assignments. Note the disappearance of cross-peaks g and j, downfield shift of cross-peaks d, e, f, h and i to give new peaks d*, e*, f*, h* and i*, respectively, and decrease in intensity of cross-peaks c, d and f after ruthenation of G3N7 (Ru-IIa) and G6N7 (Ru-IIb). For DNA sequence, see Scheme 1.](/image/article/2010/SC/c0sc00175a/c0sc00175a-f4.gif) | ||
| Fig. 4 2D [1H, 31P] HSQC NMR spectra of (A) duplex II and (B) 1.1:1 1/II mixture (0.34 mM, 0.1 M NaClO4 at 283 K, pH 7.0) in 90% H2O/10% D2O, showing the backbone 31P (−1.4 to −0.60 ppm) to sugar ring H3′ (5.2–4.6 ppm) and H4′ (4.6–4.0 ppm) connectivities. The circles indicate Cn-31P/H4′, Cn-31P/Cn−1-H3′ and Cn-31P/Gn−1-H3′ or Gn-31P/H4′, Gn-31P/Gn−1-H3′ and Gn-31P/Cn−1-H3′ assignments. Note the disappearance of cross-peaks g and j, downfield shift of cross-peaks d, e, f, h and i to give new peaks d*, e*, f*, h* and i*, respectively, and decrease in intensity of cross-peaks c, d and f after ruthenation of G3N7 (Ru-IIa) and G6N7 (Ru-IIb). For DNA sequence, see Scheme 1. | ||
2D [1H, 1H], 15N-edited [1H, 1H] NOESY NMR of products from 1.1:1 1/II reaction
Assignments for 1H NMR peaks of mono-ruthenated duplexes Ru-IIa and Ru-IIb in the spectra of the 1.1:1 1/II reaction are listed in Tables 1, S3 and S4,† and intermolecular NOEs in Tables S6 and S7.† For Ru-IIa, a large low-field shift of the G3H8 resonance was observed, as was also the case for H8 of the neighbouring G2 base and H5 and H6 of the neighbouring C4 base, relative to free duplex II (Fig. S4 and Tables S2 and S3†). The largest changes in deoxyribose H1′ chemical shifts occur for G3*, G2 and C5 residues, with the smallest changes for the neighbouring C4 and C10 residues (Fig. S4 and Table S3†). NOE cross-peaks were found between G3*H8 and 1′-en-NHd, 1′-en-NHu, H2,3, H1,4 and H9,10 protons, between G3*H1′, G3*H2′/H2′′ and 1′-H9,10 and H1,4 protons, between C4-H5′, C4-H6 and 1′-H9,10 and H1,4 protons, and between C4-H1′ and 1′-H9,10, H5,8 protons (Figs. 5, S6 and Table S6†). NOE cross-peaks were also found between protons of bases G9, G2 and C10 and 1′. In particular, NOE cross-peaks were observed between G9H2′′, C10-H1′ and 1′-H6,7 (Figs. 5, S6 and Table S6†). Sequential connectivities for base-to-sugar 1H NMR resonances were obtained, but those in the G2-C3*, G3*-C4 and G9-C10 steps were extremely weak or absent. The interruption or weakening of NOE connectivities between sequential DNA nucleotides is consistent with the binding of {(η6-tha)Ru(en)}2+ (1′) at G3* in the adduct Ru-IIa.
![Part of the 2D [1H, 1H] NOESY NMR spectrum of the 1 : 1 equilibrium mixture of duplex II and complex 1 (0.34 mM, 0.1 M NaClO4, 90% H2O/10% D2O at 283 K, pH 7.0, mixing time 400 ms). Cross-peaks: a, G9H1′/1′-H6,7; b; C10H1′/1′-H6,7; c, G8H1′/1′-H6,7; d, C7H1′/1′-H6,7. The observed intermolecular {(η6-tha)Ru(en)}2+-II cross-peaks from mono-ruthenated product II-Ru-G3 are: G3*H8/1′-enNHu, G3*H8/1′-enNHd, G3*H8/1′-H9/10, G3*H8/1′-H1,4, G3*H1′/1′-H9/10, G3*H1′/1′-H1,4, C4H6/1′-H1,4, C4H6/1′-H9,10, C4H5/1′-H9,10, G9H1′/1′-H6,7, C10H1′/1′-H6,7; and from II-Ru-G6 are: G6*H8/1′-enNHu, G6*H8/1′-enNHd, G6*H8/1′-H9/10, G6*H8/1′-H1,4, G6*H1′/1′-H9/10, C5H6/1′-H1,4, G8H1′/1′-H6,7, C7H1′/1′-H6,7. Cross-peaks within the ruthenated guanine residues G3* or G6*, and within the bound ruthenium complex 1′ are also indicated. Labels: 1′ = {(η6-tha)Ru(en)}2+; ruthenated guanines are marked with asterisks. For NMR chemical shifts, see Tables S2–S4, and for atom labels, see Fig. 1.](/image/article/2010/SC/c0sc00175a/c0sc00175a-f5.gif) | ||
| Fig. 5 Part of the 2D [1H, 1H] NOESY NMR spectrum of the 1:1 equilibrium mixture of duplex II and complex 1 (0.34 mM, 0.1 M NaClO4, 90% H2O/10% D2O at 283 K, pH 7.0, mixing time 400 ms). Cross-peaks: a, G9H1′/1′-H6,7; b; C10H1′/1′-H6,7; c, G8H1′/1′-H6,7; d, C7H1′/1′-H6,7. The observed intermolecular {(η6-tha)Ru(en)}2+-II cross-peaks from mono-ruthenated product II-Ru-G3 are: G3*H8/1′-enNHu, G3*H8/1′-enNHd, G3*H8/1′-H9/10, G3*H8/1′-H1,4, G3*H1′/1′-H9/10, G3*H1′/1′-H1,4, C4H6/1′-H1,4, C4H6/1′-H9,10, C4H5/1′-H9,10, G9H1′/1′-H6,7, C10H1′/1′-H6,7; and from II-Ru-G6 are: G6*H8/1′-enNHu, G6*H8/1′-enNHd, G6*H8/1′-H9/10, G6*H8/1′-H1,4, G6*H1′/1′-H9/10, C5H6/1′-H1,4, G8H1′/1′-H6,7, C7H1′/1′-H6,7. Cross-peaks within the ruthenated guanine residues G3* or G6*, and within the bound ruthenium complex 1′ are also indicated. Labels: 1′ = {(η6-tha)Ru(en)}2+; ruthenated guanines are marked with asterisks. For NMR chemical shifts, see Tables S2–S4,† and for atom labels, see Fig. 1. | ||
For adduct Ru-IIb, large low-field shifts were observed for the G6*H8 resonance and for H5 and H6 resonances of the neighbouring C5 residue (Fig. S5 and Tables S2 and S4†). The H6 resonance of C7 in the complementary strand, which is paired with G6, shifted slightly to low field, but the H5 resonance shifted to high-field relative to free duplex II. The largest changes in H1′ chemical shifts were found for G8, and for C7, C5, and G6*. NOE cross-peaks were found between G6*H8 and 1′-en-NHd, 1′-en NHu, H2,3, H1,4, H9,10 and H5,8 protons, between G6*-H1′ and 1′-H9,10, H5,8 and H6,7 protons, and between G6*-H4′, G6*-H5′ and 1′-H9,10 and H5,8 protons (Figs. 5, S6 and Table S7†). NOE cross-peaks were also detected between protons of the bases C5, G8, C7 and bound fragment 1′ (Figs. 5, S6 and Table S7†). Particularly of note were cross-peaks observed between C7-H2′ and 1′-H6,7, C7-H2′′ and 1′- H5,8. Sequential base-to-sugar connectivities were obtained, but those in the C4-C5, C5-G6* and C7-G8 steps were extremely weak or absent. The interruption or weakening of NOE connectivities between sequential DNA nucleotides is consistent with the binding of 1′ at G6* in the adduct Ru-IIb.
Only one set of signals was observed for the bound fragment {(η6-tha)Ru(en)}2+ (1′) in the two ruthenated duplexes Ru-IIa and Ru-IIb (Figs. 5, S6† and Table 1). Compared to the unbound chloro form of 1, peaks for 1′-H1/H4 and H2/H3 of the coordinated arene (see Fig. 1 for labelling) were shifted to low-field, the largest shift being for 1′-H1/H4 (Table 1). Peaks for 1′-H9/H10, H5/H8 and H6/H7 of the non-coordinated rings were shifted to low-field by +1.00, +0.12 and +0.09 ppm, respectively, the largest shift being for 1′-H9/H10 (△δ = +1.00 ppm). Two sets of slightly low-field-shifted or unchanged signals for 1′-en CH2 of both Ru-IIa and Ru-IIb were detected. Two sets of signals for both 1′-en-NHd and en-NHu protons of Ru-IIa and Ru-IIb were observed, and the peaks for 1′-NHd and NHu were shifted to low-field, the largest shift being for 1′-NHu. One set of signals from unreacted ruthenium complex 1 was observed in the 1.1:1 1/II reaction mixture (Fig. S7† and Table 1). These results are consistent with ROESY experiments (data not shown).
Discussion
Complex 1 selectively ruthenates guanine bases in single strand DNA I or duplex II (Fig. 2a–c) with a similar pattern to that observed for the biphenyl (bip) and p-cymene (cym) complexes.22 Complex 1 is as reactive towards duplex II as the Ru-bip complex, and much more reactive than the Ru-cym complex. This pattern of reactivity was observed previously with calf thymus DNA, for which t50% values of 10 min, 10 min and 3.5 h for complexes 1, Ru-bip and Ru-cym, respectively, were found.26Precipitation of adducts was observed when >1 mol equiv. of Ru-bip or Ru-cym complex was added to duplex II (0.2 mM).22 However, no such behaviour was observed in the present work. Addition of up to 3 mol equiv. of complex 1 to duplex II, even at the higher concentration of 0.3 mM, did not result in precipitation. This suggests that the nature of the arene influences intermolecular interactions. However, precipitation of adducts was observed when >3 mol equiv. of complex 1 was added to duplex II (0.3 mM), and was also the case when the reaction mixture of 1 + II (3:1) was kept at 277 K for long periods (ca. four weeks). Intermolecular interactions are probably also influenced by the order of occupation of the Ru sites and the extent of arene intercalation (for tha and bip).
Determination of binding sites by NMR
The 31P chemical shift changes determined from the 2D [1H, 31P] HSQC NMR experiment are consistent with ruthenation at N7 of the G residues of the 6-mer DNA duplex II by 1. Binding of Ru-bip to the phosphate of 5′-GMP27,28 causes a low-field shift of the 31P NMR resonance by up to +5.11 ppm. However, the binding of Ru-bip to N7 of 5′-GMP,28 5′-IMP or 5′-cGMP caused low-field 31P NMR shifts of less than 1 ppm. Similarly, ruthenation of 5′-GMP by trans-[RuCl2(DMSO)4] giving Ru–OPO3 coordination causes a +5.8 ppm 31P downfield shift,62 and direct Pt–OPO3 binding to IMP produces a 31P downfield shift of about +3.5 ppm.63 The formation of N7-ruthenated complexes of 5′-GMP and 5′-IMP, N6- or N4-ruthenated complexes of 5′-AMP or 5′-CMP by {Ru(III)(NH3)5}3+ gave rise to little change in 31P resonances of the nucleotides.64 Therefore, it is evident that direct coordination of RuII to a phosphate oxygen induces a 31P chemical shift change of ca. +5 ppm, while coordination to GN7 and no direct binding to phosphate oxygen induces a chemical shift change in the range of 0–1 ppm. In the present case, the most affected signals are assigned to the phosphate groups of residue C4 (Δδ +0.09 ppm) and residue G6* (Δδ −0.05 ppm) (Fig. 4 and Tables S3 and S4).† Other 31P resonances are shifted by less than 0.03 ppm. Significant changes were observed for 31P, H3′ and H4′ resonances of residues G3*, C4, C5 and G6*; minor changes observed for the corresponding resonances of C1/C7 and G2/G8 further indicated that the binding site was N7 of G3* or G6*. Selective binding to N7 of the G residues of double-stranded DNA duplex II was evident from the 15N-edited [1H, 1H] NOESY NMR spectrum and confirmed by 1H NMR chemical shift changes (Figs. 5, S6 and Tables S3 and S5†). NOE connectivities between ruthenated G*H8 and 1′-NHu or 1′-NHd were observed in the 15N-edited [1H, 1H] NOESY and 15N-decoupled [1H, 1H] NOESY NMR spectra. Binding of 1 to 9-ethylguanine and 5′-GMP27via N7 causes a low-field shift of the H8 1H NMR resonance by up to +0.6 ppm. Binding of Ru-cym and Ru-bip complexes to 6-mer single strand DNA I or duplex II23via N7 causes low-field shifts of the H8 1H NMR resonance of +0.49 to +0.66 ppm, and +0.28 to +0.58 ppm, respectively. Similar shifts were observed for the H8 resonances of G bases in the hexamer, and allow assignment of the binding sites as G3* (△δ H8 + 0.59 ppm) in Ru-IIa and G6* (△δ H8 + 0.46 ppm) in Ru-IIb present in the 1.1:1 reaction mixture of 1 + II (Figs. S1, S4 and S5, Tables S3 and S4†). With the binding fragment {(η6-tha)Ru(en)}2+ (1′), the mono-ruthenated duplex Ru-IIa is assigned as II-Ru-G3(1′), Ru-IIb as II-Ru-G6(1′) (for DNA sequence, see Scheme 1).
The ruthenation of duplex II by complex 1 mainly caused low-field shifts of imino proton resonances of G residues G3* and G9 in II-Ru-G3(1′), but high field shifts of imino proton resonances of G residues G6* and G12 in II-Ru-G6(1′) (Fig. S2 and Tables S3 and S4†). In contrast, the imino proton resonances of the mono-intercalated duplexes and di-intercalated duplex ruthenated with Ru-bip are broad and weak, implying that the base-pairs are disrupted in the duplex with an increase in dynamic mobility of the bases.23 High-field shifts of imino proton resonances were found for mono-ruthenated species in the 1:1 reaction mixture of Ru-cym complex + II, and platination of the 14-mer duplex d(TATGTACCATGTAT)/d(ATACATGGTACATA) also causes high field shifts of G imino proton resonances.23,59
Structural perturbations induced by ruthenation with complex 1 are larger than those observed for Ru-bip and Ru-cym complexes,23 and are localized to within a few (±2) base-pairs of the ruthenation site in all cases for complex 1, while only the two adjacent bases (C4 and C10 or C5 and C7) are affected by ruthenation at G3* or G6* in all cases for Ru-bip and Ru-cym adducts. Not only were large low-field shifts of the H5 and H6 resonances observed for C4 in II-Ru-G3(1′) and C5 in II-Ru-G6(1′), but also for H1′ of G2, G3, C4, C5 and C11 in II-Ru-G3(1′), and of C5, C7 and G8 in II-Ru-G6(1′) (Tables S3 and S4†).
Intercalation
Literature reports show that intercalation into DNA base pairs can often be recognised by distinctive features,19,22,23,49,54,61,65 including (a) upfield 1H NMR shifts of resonances of the intercalator; (b) NOE cross-peaks between protons of the intercalator and DNA bases at sites of intercalation; (c) the interruption or weakening of NOE connectivities between sequential DNA nucleotides; (d) the absence or weakening of the correlation peaks of H3′n-31Pn+1 and H3′n-31Pn at sites of intercalation steps and the large chemical shift perturbations at the intercalation steps; and (e) the weakening of the strength of H-bonding between en-NH and GO6 in the case when the ruthenium complex Ru-bip with an extended arene ring system was involved.22,23It is notable that no large high-field shifts of proton resonances of 1′ were detected, but large low-field shifts up to +1.00 ppm were observed for protons H9,10, H5,8 and H6,7 of rings B and C in the mono-ruthenated duplexes II-Ru-G3(1′) and II-Ru-G6(1′) (Table 1). These shifts are inconsistent with shielding effects from the ring-currents of nucleobases which form a sandwich with the intercalated non-coordinated rings of bound 1′, and so do not provide evidence for intercalative binding.19,49,54,65 For example, upfield shifts of between −0.4 and −1.0 ppm have been reported for Ru-bip intercalated into 6-mer or 14-mer duplex DNA,22,23 and upfield shifts of −0.1 to −1.0 ppm for the intercalated dap (1,12-diazaperylene) ligand of the dirhodium(II) carboxylate complex [Rh2(dap)(CH3COO)3(CH3OH)3] into a 12-mer duplex DNA.19 Such large low-field shifts of the bulky tha intercalator have not been observed for other bulky intercalators, for example, large high-field shifts have been observed for bulky intercalated cholesterol groups.44 However, similar large low-field shifts for proton resonances of 1′-9EtG were found for the adduct [(η6-tha)Ru(en)(9EtG)];28 the H5,8 and H6,7 resonances slightly shifted to high-field, but the H9,10 resonances remained unchanged. In the case of [(η6-tha)Ru(en)(5′-GMP)] (1′-GMP),28 H9,10 and H5,8 resonances are shifted to low-field by +0.83 and +0.50 ppm, respectively, and the H6,7 resonances shifted to high-field by −0.10 ppm. In the present case of the mono-ruthenated duplexes II-Ru-G3(1′) and II-Ru-G6(1′), H9,10, H5,8 and H6,7 resonances shifted to low-field by +1.00, +0.12 and +0.09 ppm, respectively. Thus it is reasonable that the resonances of intercalated non-aromatic rings B and C of tha in the mono-ruthenated duplexes II-Ru-G3(1′) and II-Ru-G6(1′) shift to low field.
The single crystal X-ray structure of (1′-GMP)28 shows that ring C of 1′ is tilted towards the purine by 27.8° and lies directly over the purine base, indicative of strong intramolecular π–π stacking between ring C and the purine ring with a centroid–centroid separation of 3.45 Å and dihedral angle of 3.3°. Intercalation of the non-coordinated rings of 1′ into the DNA duplex is also consistent with circular and linear dichroism data.26,29 Due to excessive resonance broadening, the resonances for protons that intercalate between purine rings are difficult to assign. Weak to intermediate intensity NOE cross-peaks were found not only between the rings of bound 1′ and H1′ or H8 protons of G3* or C4 in II-Ru-G3(1′), but also between the rings of 1′ and G9 and C10 (Figs. 5, S6 and Table S6†). This can happen if the intercalation occurs not only at the G3pC4 base step, but also at the G9pC10 base step. The intermediate intensity cross-peaks observed between G9H2′′ or C10H1′ and 1′-H6,7 protons, indicate that the extended rings of 1′ intercalate deeply and are located between the middle of G9 and C10 bases. Analogous NOE cross-peaks between the rings of bound 1′ and H1′, H8, H2′ and H2′′ of G6* in II-Ru-G6(1′), and also between rings of 1′ and C5, C7 and G8 were detected, indicating that intercalation occurs between G6* and C5, and between G8 and C7 as well (Figs. 5 and S6, and Table S7†). The intermediate intensity cross-peaks observed between G6 or C5 and 1′-H1,4 protons, indicate that the coordinated arene ring of 1′ is located between the middle of G6 and C5 bases. Additionally, intermediate intensity cross-peaks observed between C7H2′ and 1′-H6,7 and C7H2′′ and 1′-H5,8 protons indicate that the extended rings of 1′ intercalate deeply and are located near to the C7 base. The interruption of NOE connectivity pathways between the corresponding base pairs (G2-C3*, G3*-C4 and G9-C10 steps in II-Ru-G3(1′), and C4-C5, C5-G6* and C7-G8 steps II-Ru-G6(1′)) is consistent with these intercalation sites.
The absence of the H3′n-Pn+1 cross-peaks linking the C5-G6* step and the H3′-P cross-peaks of G3*, C4 and C5, the low field shifts for H3′n-Pn+1 cross-peaks linking G2-G3*, C4-C5 and G3*-C4, and for H3′-P cross-peaks of C4/C10 and G6, and the large chemical shift perturbations at the G2-G3* and C5-G6* steps, together indicate that the intercalation occurs between G3pC4 or C5pG6 base steps (Fig. 4).62 Previous work has shown that the intercalation sites of the non-coordinated phenyl ring of Ru-bip in mono-ruthenated duplexes4c are also between G3pC4 or C5pG6 base steps.
No cross-peaks for en-NHu resonances of Ru-IIa and Ru-IIb were detected after the 1.1:1 1/II reaction mixture had been freeze-dried and re-dissolved in D2O. This suggests that the hydrogen bond between G*O6 and en-NH of 1′ is weakened (Fig. S3 and Table S5†), which is consistent with intercalation of the non-coordinated rings of 1′ into duplex DNA II. Similarly weakened hydrogen bonds were also observed when the biphenyl ring of Ru-bip intercalates into the hexamer duplex.22 The strength of the H-bond between G*O6 and en-NH is related to the decay rate of the en-NH signals when II-Ru adducts are dissolved in 99% D2O.22 For the non-intercalated adduct with Ru-cym, the half-life was 72 h. However, those of the mono- and di-intercalated Ru-bip adducts were only 5 h and <0.1 h, respectively.
NMR studies show that the arene–nucleobase π–π stacking of 1′ with hexamer duplex is different from that of Ru-bip.22,23 Only a very few weak NOE contacts between protons of ring B of biphenyl and H1′ and H2′/H2′′ protons of G3* or C4 of the hexamer duplex are observed.22 The protons Ho′, Hp′ and Hm′ of bound Ru-bip in the DNA duplex adducts were consistently shielded relative to free Ru-bip, consistent with base stacking of the non-coordinated ring B between base pair G3*pC4. However, in the present case, not only were weak to intermediate intensity NOE contacts detected between protons of rings B and C of tha and protons of G3* and C4 or G6* and C5, but also intermediate intensity NOE contacts were detected between protons of ring C of tha and protons of G9 and C10 or C7 and G8 DNA bases which pair with G3* or C4, G6* or C5 in the complementary DNA strand (Fig. 5, Tables S6 and S7†), respectively. This indicates that rings B and C of tha are involved in a penetrative intercalation between two pairs of bases, G3/C10:C4/G9 or G6/C7:C5/G8. It is interesting that large low-field shifts, but not large high-field shifts are observed for the proton resonances of intercalated rings B and C of 1′, indicating that the intercalative interactions between Ru-tha and Ru-bip with the DNA duplex are significantly different from one another. The ring current shifts are position-related: upfield shifts only arise when the protons are above or below the ring plane. In contrast, resonances for protons located close to the plane and beyond the confines of the ring are shifted to low field.66
Modelling of II-Ru-G3(1′) and II-Ru-G6(1′)
In order to attempt to rationalize the NOE and chemical shift information gathered for II-Ru-G3(1′) and II-Ru-G6(1′), two molecular models were constructed in which the duplex d(CGGCCG)2 was ruthenated at N7 of G3 or G6 by 1′, as shown in Fig. 6. The II-Ru-G3(1′) (Fig. 6a–c) and II-Ru-G6(1′) (Fig. 6d–f) models are mostly consistent with the NMR observations, and overwhelming evidence indicates that a penetrative interaction occurs between rings B and C of 1′ and duplex DNA at G3/C10:C4/G9 or G6/C7:C5/G8, respectively. The model also provides a useful indication of how 1′ may lie in relation to its DNA binding site. The H5,8 and H9,10 protons of 1′ are located above or below the ring plane and the distances between H5,8 or H9,10 protons and purine or pyrimidine rings of G or C bases are nearly 0.9 Å shorter than the case of aromatic intercalators, such as Ru-bip. Compared to the structural findings for aromatic intercalators, both structures (II-Ru-G3(1′) and II-Ru-G6(1′)) suggest distortion of bases-pair planes around the site at which the tha has penetrated and the system is likely to be highly dynamic in and around the intercalation sites. The dynamics of II-Ru-G6(1′), in which the Ru binds to a terminal, potentially frayed nucleotide, would be expected to be significantly different compared with those for II-Ru-G3(1′), in which the Ru binds to the internal base.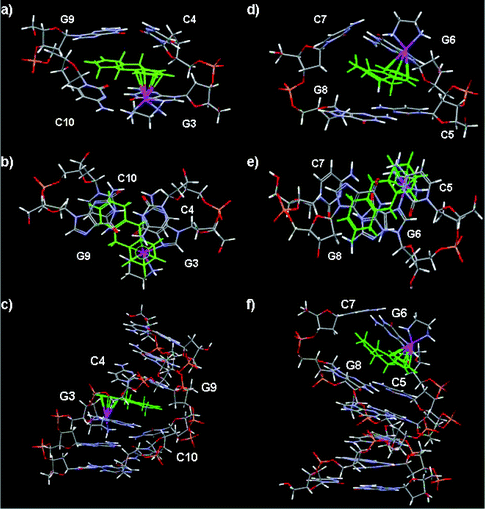 | ||
| Fig. 6 Molecular models of duplex II ruthenated at N7 of G3 or N7 of G6 with {(η6-tha)Ru(en)}2+. (a)–(c) II-Ru-G3 showing the intercalation of the tetrahydroanthracene ligand between G3/C10:C4/G9. (d)–(f) II-Ru-G6 in which the non-arene rings are intercalated between G6/C7:C5/G8. In each case side and top views of the intercalation site are shown as well as the whole duplex (bottom). Colour code: tha green, Ru purple, P yellow, O red. | ||
The data imply that in both cases the tha of 1′ in the models has swung round so that the ring system points across to the opposite strand rather than penetrating so deeply into the strand to which the Ru centre is attached (G3* or G6*). This supports the NOE contacts observed between protons of rings A and B of 1′ and H1′ and H8 or H6 protons of G3, C4 or C5, G6 and in particular between H6,7 protons of ring C of 1′ and the H1′, and H2′ protons of residue C10, G9 or G8, C7 of the complementary strand (see Tables S6 and S7,†Fig. 6). In contrast in models for Ru-bip, the non-coordinated phenyl ring of the biphenyl ligand penetrates deeply at the G3*pC4 or G6*pC5 base step. The relevant inter-proton distances are consistent with the observed NOE contacts. Observed NOE data are consistent with the model of II-Ru-G3(1′): NOEs occur between H1,4 (∼3.45 Å), H9,10 (∼4.73 Å), H2,3 (∼3.72 Å) and G3*H8, respectively; between H9,10 (∼4.74 Å) or H1,4 (∼4.64 Å) and G3*H1′, respectively; between H9,10 (∼3.71 Å) or H1,4 (∼3.24 Å) and C4H6, H5,8 (∼3.60 Å) or H9,10 (∼4.14 Å) and C4H1′, respectively; between H6,7 (∼3.67, 3.18 or 2.52 Å) and G9H1′, -H2′ or -H2′′, respectively; between H6,7 (∼2.94 or 2.69 Å), H5,8 (∼3.08 Å) and C10H6 or C10H1′, respectively and NHd (∼2.49 or 3.03 Å) and G2H8 or -H2′ in the model, respectively. Observed NOE data are also consistent with the model II-Ru-G6(1′): medium and weak NOEs occur between H1,4 (∼2.74 Å) or H9,10 (∼2.86 Å) and G6*H8, respectively; between H5,8 (∼2.55 Å), H9,10 (∼3.47 Å) and G6*H1′, respectively; between H1,4 (∼2.87 Å) and C5H6, H1,4 (∼3.72 Å) and C5H1′, H1,4 (∼2.66 Å) and C5H2′, respectively; between H6,7 (∼2.44 Å) and C7H2′, H6,7 (∼2.94 Å) and C7H1′, H6,7 (∼3.01 Å), H5,8 (∼2.53 Å) and C7H2′′, H6,7 (∼3.44 Å), H5,8 (∼3.50 Å) and G8H1′, H5,8 (∼3.85 Å), H6,7 (∼3.89 Å) and G8H2′/H2′′ in the model, respectively. As shown in Fig. 6, the shortest distance for enNH⋯O6G3 or enNH⋯O6G6 is 2.66 or 2.01 Å, larger than that in the reported 9EtG adduct (1.91 Å),28 consistent with weak en-NH resonances observed for II-Ru-G3(1′) and II-Ru-G6(1′) in D2O.
The largest downfield shift (+1.00 ppm) is observed for H9,10 protons of ring B of 1′ for mono-ruthenated duplexes II-Ru-G3(1′) and II-Ru-G6(1′) (Table 1), but the shift changes for H6,7 (+0.09 ppm) and H5,8 (+0.12 ppm) of ring C of 1′ are rather small when compared with that for H9,10. These results are consistent with the base stacking of the non-coordinated rings and formation of short C–H⋯X (X = O or N)67 hydrogen bonds between the protons of non-coordinated rings and bases as shown in models II-Ru-G3(1′) and II-Ru-G6(1′) (Fig. 6). It is clear that H5,8 and H6,7 protons are located within the confines of the purine ring G9 in model II-Ru-G3(1′) or G6 in II-Ru-G6(1′), and the H9,10 protons are located exactly in the middle of the two strands (Fig. 6) and in the ring planes beyond the confines of the purine or pyrimidine rings.
It has been reported that proton chemical shifts may change by up to +2.1 ppm (downfield) on formation of C–H⋯X (X = O or N) hydrogen bonds.68 It is clear in the present case that the protons of rings B and C do sit near the edge of the purine or pyrimidine rings from the models II-Ru-G3(1′) and II-Ru-G6(1′), and the protons are not directed to the centres of these rings. Other than the fact that protons of ring B are within the arene ring plane in Ru-bip, the H5,8 and H9,10 protons of 1′ are located above or below the ring plane in the models. Such orientation makes the distances between H5,8 or H9,10 and N or O atoms of purine or pyrimidine rings of G or C bases nearly 0.9 Å shorter than those of Ru-bip. For model II-Ru-G3(1′): short H9,10⋯N1 of G3 (∼2.68 Å), H9,10⋯N3 of C4 (∼2.51 Å), H5,8⋯N1 or N7 of G9 (∼2.70 or 2.75 Å, respectively), H5,8⋯OC2 of C4 (∼2.60 Å), H6,7⋯sugar O of G9 (∼2.58 or 2.80 Å, respectively) distances are observed (Fig. 6a–c). For model II-Ru-G6(1′): short H9,10⋯N9 of G6 (∼2.66 Å), H9,10⋯N3 of C5 (∼2.54 Å), H5,8⋯N3 of G6, C7 and G8 (∼2.60, 2.61 or 2.61 Å, respectively), H6,7⋯OC2 of C7 (∼2.59 Å) distances are observed (Fig. 6d–f). Thus, in the present case, the shifts of the protons of rings B and C of 1′ reflect both the downfield shift induced by the formation of C–H⋯X (X = O or N) hydrogen bonding,68 and the upfield shift induced by the intercalation effect on the protons located above or below the intercalator, due to the ring current effect of the aromatic groups. The H9,10 protons are located exactly central to the two strands of DNA, so the downfield shifts for H9,10 protons are larger (1.0 ppm).
The data imply that in both cases the tha of 1′ in the models has swung round so that the ring system points across to the opposite strand rather than penetrating so deeply into the strand to which the Ru centre is attached (G3* or G6*). This tendency is then likely to be stabilized by tha–base interactions and C–H⋯X (X = O or N) hydrogen bonding within the G3-tha ring-C4 and the G9-tha ring-C10 ‘sandwich’ as shown in Fig. 6a–c, or within the G6-tha ring-C5 and the G8-tha ring-C7 ‘sandwich’ as shown in Fig. 6d–f. This kind of intercalation distorts the DNA more than that of aromatic intercalators, such as Ru-bip, and reduces the strength of H-bonding between en-NH and G3O6. The intercalation of the non-coordinated phenyl ring of Ru-bip,22 of actinomycin D (ActD) and daunomycin65 between the GC step in previous work suggested that steric crowding at the GpC step is less than that at the GpG step, which thereby allows accommodation of the bulky non-aromatic rings of tha. A further driving force for GpC rather GpG intercalation is the weaker purine–pyrimidine π–π stacking interaction for GpC compared with purine–purine GpG steps.65 It is interesting in the present work that all of the intercalation of Ru-bound tetrahydroanthracene occurs between GpC base steps, and there is no evidence for intercalation at the GpG base steps.
Ru-en-NH⋯GO6 H-bonding
Two distinct NHu-NHd cross-peaks were identifiable in the 283 K 2D NOESY and 2D 15N-edited NOESY NMR spectra (Table S5 and Figs. 5, S6†), and the downfield shifts of NH resonances in the adducts are consistent with the presence of H-bonding to the C6 carbonyl of the coordinated G.22,23,27,28 Only one broad peak for NH(u) was detected in the 2D [1H, 15N] HSQC NMR data at 283 K; the resonance for NH(d) was too broad to observe (Fig. S3†). NH exchange was rapid for the en-NH(u) protons in the mono-intercalated duplexes II-Ru-G3(1′) and II-Ru-G6(1′) (no 1H/15N cross-peaks being detected for a D2O solution of II-Ru-G3(1′) and II-Ru-G6(1′), Fig. S3†). Such a rapid NH exchange of the en-NH(u) protons was also observed in the double-intercalated duplex II-Ru2-G3G9(Ru-bip),22 suggesting that mono-intercalation of 1′ into duplex II gives rise to similar effects to that of the di-intercalation of Ru-bip.Sequence specificity of G metallation
Adducts of duplex II eluted as single strands from the reverse-phase HPLC column22,23 (Fig. 2d and 2e) due to the denaturing character of the HPLC solvent. Three mono-ruthenated products (I-Ru-G2, I-Ru-G3 and I-Ru-G6) and three di-ruthenated products (I-Ru2-G2G3, I-Ru2-G2G6 and I-Ru2-G3G6) were detected in the 1:1 1/I reaction mixture, showing that all three guanine bases can be ruthenated readily in the single-stranded hexamer DNA d(CGGCCG) (Table S1†). Only the two mono-ruthenated products II-Ru-G3 and II-Ru-G6 were detected in the reaction of duplex II with 1 at a Ru:II molar ratio of 1:1; no G2 ruthenated adduct was detected (Table S1†). When the above mono-ruthenated products II-Ru-G3 and II-Ru-G6 were reacted with a second mol equiv. of 1 at the same temperature, only two mono-ruthenated single strand adducts (I-Ru-G3 and I-Ru-G6) and two di-ruthenated single strand adducts (likely to be I-Ru2-G3G6 and I-Ru2-G2G6) eluted from the reverse-phase HPLC column (Table S1†). The products may therefore involve ruthenation on the same strand: II-Ru2-G3G6 and II-Ru2-G2G6, or on different strands: II-Ru2-G3G9, II-Ru2-G6G9, II-Ru2-G6G12 and II-Ru2-G3G12. No G8 ruthenated duplex adducts such as II-Ru2-G3G8 or II-Ru2-G6G8 were detected. These results are consistent with 2D TOCSY experiments (Fig. 3): the cross-peak intensities of C-H5/H6 (C1′, C4′ and C5′) of ruthenated species increased in the 2:1 1/II reaction mixture.
The cross-peaks for H5/H6 resonances of C5/C11 almost disappeared when the Ru:II ratio was raised to 3:1 (Fig. 3D). This might suggest that the third ruthenation site for the di-ruthenated duplexes occurs at the un-ruthenated G6 and G12 residues, to form two tri-ruthenated duplex adducts: II-Ru3-G3G6G12 and II-Ru3-G2G6G12. Thus, ruthenation of mono-ruthenated products II-Ru-G3 and II-Ru-G6 might result in three di-ruthenated duplex species: II-Ru2-G3G6, II-Ru2-G2G6 and II-Ru2-G6G9 (Scheme 1, Table S1†). The HPLC, MS and 2D 15N-decoupled [1H, 1H] TOCSY NMR data indicate that the selectivity of G base ruthenation for the free duplex II, mono-ruthenated duplexes and di-ruthenated duplexes is quite different. For free duplex II, little ruthenation of G2 was observed; for mono-ruthenated duplexes II-Ru-G3 and II-Ru-G6, little ruthenation of G8 was observed; however, the favoured ruthenation site for the di-ruthenated duplexes appears to be G6 and G12, the G bases at the 3′ end. Reactions of complexes Ru-cym and Ru-bip with duplex II d(CGGCCG)2 at a Ru:II ratio of 1:1, also gave rise to little ruthenation of G2.22 However, the II-Ru-G3:II-Ru-G6 ratios in the reaction mixtures with Ru:II ratio of 1:1 are different: 1:1 for Ru-cym, 3:1 for Ru-bip and 2:1 for Ru-tha in the present case, indicating that there is no preference for binding to an internal base or terminal nucleotide for the non-intercalator Ru-cym. In contrast, obvious specificity exists for binding to an internal base for the aromatic intercalator Ru-bip. Specificity for internal bases is also the case for the non-aromatic intercalator Ru-tha. Exclusive attack on the 3′-G (G3), as seen for these organometallic Ru arene complexes, is uncommon for platination. There might be two reasons for this. Firstly, the pseudo-octahedral coordination site on an arene RuII complex is more sterically demanding than that of a square-planar site on PtII. In addition, the steric hindrance around each base in DNA sequences is quite different. The combined steric hindrance of G and C plus the RuII complex are likely to account for a preferential binding to G3N7 or G6N7 rather than to G2N7 in the free duplex. After ruthenation at G3 or G6, the situation for the non-ruthenated single strand in the mono-ruthenated products II-Ru-G3 and II-Ru-G6 may be similar to that of the free duplex, so no G8 ruthenation is detected. For the mono-ruthenated single strand I-Ru-G3 or I-Ru-G6 in the mono-ruthenated products II-Ru-G3 and II-Ru-G6, steric hindrance around G2 in the mono-ruthenated duplex II-Ru-G3 is much greater than that in the free duplex and for this reason it is believed that no II-Ru2-G2G3 species is detected. The DNA distortion caused by ruthenation at N7 of G6 may decrease the steric hindrance around G2 in the mono-ruthenated duplex II-Ru-G6, resulting in formation of the G2-bound di-ruthenated duplex II-Ru2-G2G6. Steric hindrance around G2 and G3 in the di-ruthenated duplexes II-Ru2-G3G6, II-Ru2-G2G6 and II-Ru2-G6G9 is even greater, so ruthenation at the end base G6 or G12 as the third site is reasonable.
Bulky lesion and penetrative intercalation
Reported data44 show that the intercalative binding of a bulky intercalator cholesterol group on a modified base of a duplex DNA is a classical intercalation, and the lesion site and the distortions in the structure of the DNA produced by these cholesterol derivatives are somehow similar to those induced by other adducts containing polycyclic aromatic groups. However, the base opposite the modified nucleotide is displaced and the local structure of the double helix is highly distorted. These observations are quite different from those in the present work involving penetrative intercalation of tha into duplex DNA when Ru-tha is coordinated at GN7. It is notable that there is no displacement of the base opposite the modified nucleotide, but C–H⋯X (X = O or N) hydrogen bonding between protons of ring C of tha and O or N atoms of bases opposite the ruthenated nucleotides. The local structure of the ruthenated double helix is highly distorted. Such a highly distorted double helix is not observed in the DNA adducts of platinum complexes with an acridine side arm intercalator, e.g. PT-ACRAMTU,56 where the threading intercalation occurs at the central base-pair step but does not cause helical bending.Experimental section
Materials
Organometallic ruthenium(II) complex [(η6-tha)Ru(en)Cl][PF6] (1PF6) (tha = 1,4,9,10-tetrahydroanthracene, en = ethylenediamine) and 15N-labeled 1 (15N-1) were synthesised as described previously.27,28 The sodium salt of FPLC-purified oligonucleotide d(CGGCCG) I was purchased from Oswel (Southampton, UK) and was further purified by HPLC. Sodium perchlorate and acetonitrile (HPLC grade) were obtained from Fisher, and triethylammonium acetate buffer (TEAA) from Fluka.High performance liquid chromatography (HPLC)
This was carried out on reversed-phase columns with TEAA and TEAA/acetonitrile as mobile phases.HPLC-electrospray ionisation mass spectrometry (HPLC-ESIMS)
Negative-ion electrospray ionisation mass spectra were obtained on a mass spectrometer interfaced with a reversed phase HPLC column eluted with TEAA/acetonitrile gradients as above.NMR spectroscopy
NMR data were acquired on an 800 MHz or 600 MHz Bruker Avance NMR spectrometer equipped with a multiple resonance TXI (1H, 13C, 15N, 31P) xyz-gradient probe.Molecular modelling
A structure for canonical B-form duplex d(CGGCCG)2 was generated within the biopolymer module of Sybyl (version 6.3, Tripos Inc.). Crystal coordinates from the X-ray crystal structures of 1 allowed accurate representation of the Ru complex to be incorporated into the model Ru-DNA constructs. Docking of the Ru-complex onto GxN7 (x = 3 or 6) of the DNA structure was achieved by manual independent manipulation of both DNA and Ru-complex molecules. The Ru-N7 inter-atomic distance was based on reported crystal structures of Ru-GMP complexes. A pseudo-atom at the centre of the η6-six-membered aromatic ring (ring A) of tha was attached to the Ru centre to provide a rotatable bond about which the tha moiety could be manipulated. In a similar way, the Ru–GN7 bond was activated to form a rotatable bond, about which the entire Ru ligand could be rotated independently of the DNA structure. Ru–GxN7 models were prepared in such a way as to reduce steric contact as far as possible. Constraints were applied where deemed plausible and structures were energy minimized to remove the effects of steric clash.Details of reactions of II with 15N-1, HPLC, HPLC-ESI-MS, NMR and pH measurements are in the ESI.†
Conclusions
In conclusion, the results presented here provide a rare example of coordinative binding and the penetrative intercalation of a bulky intercalator into DNA, and may help to explain why ruthenium arene complexes have a different mechanism of antitumour activity (perhaps related to recognition by nucleotide repair enzymes) compared to cisplatin. Firstly, the NMR results were indicative of the penetrative intercalation of the tha rings B and C of 1′, selectively between two base pairs G3/C10:C4/G9 or G6/C7:C5/G8, which contrasts with that observed between one base pair G3/C4 and or G6/C5 for the classic aromatic intercalator Ru-bip. The two slightly different intercalation models for the extended non-aromatic rings of Ru-tha, indicate that the distortion of the DNA duplex is sequence-related. Secondly, large low-field shifts for proton resonances of the intercalated non-coordinated rings B and C of tha reflect both the downfield shift induced by the formation of short C–H⋯X (X = O or N) hydrogen bonds and upfield shift induced by the intercalation effect on the protons located above or below the intercalator, due to the ring currents of aromatic groups. The downfield shifts of H9,10 protons of 1′ are larger (+1.0 ppm) for they are located exactly in the middle of the two strands. Such deshielding of intercalator NMR resonances is rare, indicating that the intercalative interactions between this bulky tha intercalator and classical aromatic DNA intercalators are somewhat different. Thirdly, the DNA structural perturbations induced by Ru-tha are larger than those observed for Ru-bip and Ru-cym complexes; distortions of base-pair planes are observed around the sites at which the tha has penetrated, and the dynamics of the terminal base ruthenated adduct II-Ru-G6(1′) are significantly different from those of internal base ruthenated adduct II-Ru-G3(1′). These findings agree with the fact that the precipitation of DNA duplex adducts of Ru-tha is observed only at very high concentrations compared with Ru-bip, suggesting that the intercalation of sterically bulky tha into a DNA duplex makes the duplex DNA behave differently from intercalation by the aromatic bip. Fourthly, selective ruthenation at N7 of G3 and G6 in the hexamer DNA duplex is similar to that of Ru-cym and Ru-bip, but the mono-intercalation of tha reduced the strength of H-bonding between en-NH and GO6 as much as that for the di-intercalated di-ruthenated Ru-bip duplex. Intercalation at GpC by tha appears to have a lower energy penalty when compared with intercalation at GpG base steps, thereby allowing accommodation of the non-aromatic, bulky rings of tha. Although all 3 G's were readily ruthenated at N7 in the single-stranded DNA hexamer, only G3 (or G9) and G6, and not G2 (G8) were ruthenated in the free DNA duplex which is attributed to unfavorable steric interactions22 between the duplex and arene for binding at G2 (G8). The different ratios of II-Ru-G3:II-Ru-G6 adducts in the reaction mixtures with Ru:II ratio of 1:1, indicates that there are differences in specificity from binding to internal bases or terminal nucleotides for the non-intercalator Ru-cym, the aromatic intercalator Ru-bip and non-aromatic intercalator Ru-tha. Little ruthenation of G8 was observed in the mono-ruthenated duplexes, but the favorable binding sites were G6 and G12 when di-ruthenated duplexes were reacted with {(η6-tha)Ru(en)}2+. These results also demonstrate that the combination of HPLC, ESI-MS together with 2D [1H, 1H] TOCSY NMR experiments is powerful for elucidating the selectivity of G-base ruthenation of the free duplex II, mono-ruthenated duplexes and di-ruthenated duplexes. Such knowledge of DNA interactions may be incorporated into design concepts for this class of anticancer agents and assist the exploration of structure–activity relationships.
The coordinative and penetrative intercalative interactions between Ru-tha and duplex DNA are different from that of DNA modified covalently by aromatic or bulky intercalators, in which the displacement or flip-out of bases near the modified sites may occur. Although both involve the modification of a DNA base via coordinative bonding, the penetrative intercalative interactions between Ru-tha and duplex DNA are also different from that of platinum complexes with an acridine side arm intercalator, where the threading intercalation does not cause helical bending. The C–H⋯X (X = O or N) hydrogen bonds between protons of ring C of tha and O or N atoms of bases opposite the ruthenated nucleotides may contribute significantly to the intercalative interaction between Ru-tha and duplex DNA. The fact that penetrative intercalation has rarely been reported for mono-metallointercalators, implies that the direct Ru–N bonding may also assist with penetrative intercalation for the bulky tha ligand. Unwinding and distortion, while still maintaining the basic duplex structure, could contribute to the toxicity of the Ru-tha complex by hindering DNA repair. A bulky lesion is one of the six main DNA lesions that may invoke NER, for example, the first and rate-determining step in NER is the recognition of the bulky lesions by the XPC/HR23B protein heterodimer complex.69 However, mutations and potentially cancer may result if the bulky lesions are resistant to NER.70 The high anticancer activity both in vitro and in vivo and the high potency of the tha complex may arise in part from the lack of repair of the lesions formed on DNA by this complex,29 and assist with elucidation of structure–activity relationships for this class of complexes.
Acknowledgements
We thank the Wellcome Trust (Travelling Fellowship for HL), NSF (20871069) and JSSF (BK2008428) and facilities in the Edinburgh Protein Interaction Centre and Oncosense Ltd for their support for this work, Dr Haimei Chen for the gift of some of the complexes and colleagues in the EC COST Action D39 for stimulating discussions.Notes and references
- Y. K. Yan, M. Melchart, A. Habtemariam and P. J. Sadler, Chem. Commun., 2005, 4764 RSC.
- M. J. Clarke, Coord. Chem. Rev., 2003, 236, 209 CrossRef CAS.
- E. Alessio, G. Mestroni, A. Bergamo and G. Sava, Curr. Top. Med. Chem., 2004, 4, 1525 CrossRef CAS.
- P. J. Dyson, Chimia, 2007, 61, 698 CrossRef CAS.
- J. E. Debreczeni, A. N. Bullock, G. E. Atilla, D. S. Williams, H. Bregman, S. Knapp and E. Meggers, Angew. Chem., Int. Ed., 2006, 45, 1580 CrossRef CAS.
- M. A. Jakupec and B. K. Keppler, Curr. Top. Med. Chem., 2004, 4, 1575 CrossRef CAS.
- F. Schmitt, P. Govindaswamy, G. Süss-Fink, W. Han Ang, P. J. Dyson, L. Juillerat-Jeanneret and B. Therrien, J. Med. Chem., 2008, 51, 1811 CrossRef CAS.
- P. M. Takahara, A. C. Rosenzweig, C. A. Frederick and S. J. Lippard, Nature, 1995, 377, 649 CrossRef CAS.
- D. B. Zamble and S. J. Lippard, in Cisplatin – Chemistry and Biochemistry of a Leading Anticancer Drug, ed. B. Lippert, Wiley-VCH, New York, 1999, p. 73 Search PubMed.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307 CrossRef CAS.
- B. E. Bowler and S. J. Lippard, Biochemistry, 1986, 25, 3031 CrossRef CAS.
- W. J. Sundquist, D. P. Bancroft and S. J. Lippard, J. Am. Chem. Soc., 1990, 112, 1590 CrossRef CAS.
- L. Maresca, C. Pacifico, M. C. Pappadopoli and G. Natile, Inorg. Chim. Acta, 2000, 304, 274 CrossRef CAS.
- J. R. Choudhury and U. Bierbach, Nucleic Acids Res., 2005, 33, 5622 CrossRef CAS.
- K. E. Erkkila, D. T. Odom and J. K. Barton, Chem. Rev., 1999, 99, 2777 CrossRef CAS.
- L. M. Wilhelmsson, F. Westerlund, P. Lincoln and B. Norden, J. Am. Chem. Soc., 2002, 124, 12092 CrossRef CAS.
- A. Greguric, G. I. Greguric, T. W. Hambley, J. R. Aldrich-Wright and J. G. Collins, J. Chem. Soc., Dalton Trans., 2002, 849 RSC.
- U. Schatzschneider and J. K. Barton, J. Am. Chem. Soc., 2004, 126, 8630 CrossRef CAS.
- M. Kang, A. Chouai, H. T. Chifotides and K. R. Dunbar, Angew. Chem., Int. Ed., 2006, 45, 6148 CrossRef CAS.
- C. Gossens, I. Tavernelli and U. Rothlisberger, J. Am. Chem. Soc., 2008, 130, 10921 CrossRef CAS.
- P. C. A. Bruijnincx and P. J. Sadler, Curr. Opin. Chem. Biol., 2008, 12, 197 CrossRef CAS.
- H. K. Liu, F. Y. Wang, J. A. Parkinson, J. Bella and P. J. Sadler, Chem.–Eur. J., 2006, 12, 6151 CrossRef.
- H. K. Liu, S. J. Berners-Price, F. Y. Wang, J. A. Parkinson, J. J. Xu, J. Bella and P. J. Sadler, Angew. Chem., Int. Ed., 2006, 45, 8153 CrossRef CAS.
- R. E. Morris, R. E. Aird, P. D. Murdoch, H. M. Chen, J. Cummings, N. D. Hughes, S. Parsons, A. Parkin, G. Boyd, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2001, 44, 3616 CrossRef CAS.
- R. E. Aird, J. Cummings, A. A. Ritchie, M. Muir, R. E. Morris, H. M. Chen, P. J. Sadler and D. I. Jodrell, Br. J. Cancer, 2002, 86, 1652 CrossRef CAS.
- O. Novakova, H. M. Chen, O. Vrana, A. Rodger, P. J. Sadler and V. Brabec, Biochemistry, 2003, 42, 11544 CrossRef CAS.
- H. M. Chen, J. A. Parkinson, R. E. Morris and P. J. Sadler, J. Am. Chem. Soc., 2003, 125, 173 CrossRef CAS.
- H. M. Chen, J. A. Parkinson, S. Parsons, R. A. Coxall, R. O. Gould and P. J. Sadler, J. Am. Chem. Soc., 2002, 124, 3064 CrossRef CAS.
- O. Novakova, J. Kasparkova, V. Bursova, C. Hofr, M. Vojtiskova, H. M. Chen, P. J. Sadler and V. Brabec, Chem. Biol., 2005, 12, 121 CrossRef CAS.
- H. M. Chen, J. A. Parkinson, O. Novakova, J. Bella, F. Y. Wang, A. Dawson, R. Gould, S. Parsons, V. Brabec and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 14623 CrossRef CAS.
- M. Castellano-Castillo, H. Kostrhunova, V. Marini, J. Kasparkova, P. J. Sadler, J. M. Malinge and V. Brabec, J. Biol. Inorg. Chem., 2008, 13, 993 CrossRef CAS.
- C. Lian, H. Robinson and A. H. J. Wang, J. Am. Chem. Soc., 1996, 118, 8791 CrossRef CAS.
- K. W. Jennette, S. J. Lippard, G. A. Vassiliades and W. R. Bauer, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 3839 CAS.
- W. I. Sundquist and S. J. Lippard, Coord. Chem. Rev., 1990, 100, 293 CrossRef CAS.
- B. P. Hudson and J. K. Barton, J. Am. Chem. Soc., 1998, 120, 6877 CrossRef CAS.
- J. G. Collins, R. M. Rixon and J. R. Aldrich-Wright, Inorg. Chem., 2000, 39, 4377 CrossRef CAS.
- L. Gonzalez-Bulnes and J. Gallego, J. Am. Chem. Soc., 2009, 131, 7781 CrossRef CAS.
- J. Tan, N. E. Geacintov and S. Broyde, J. Am. Chem. Soc., 2000, 122, 3021 CrossRef CAS.
- A. J. Danford, D. Wang, Q. Wang, T. D. Tullius and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12311 CrossRef CAS.
- F. Coste, J.-M. Malinge, L. Serre, W. Shepard, M. Roth, M. Leng and C. Zelwer, Nucleic Acids Res., 1999, 27, 1837 CrossRef CAS.
- Z. Johar, A. Zahn, C. J. Leumann and B. Jaun, Chem.–Eur. J., 2008, 14, 1080 CrossRef CAS.
- N. E. Geacintov, M. Cosman, B. E. Hingerty, S. Amin, S. Broyde and D. J. Patel, Chem. Res. Toxicol., 1997, 10, 111 CrossRef CAS.
- M. Lukin and C. de Los Santos, Chem. Rev., 2006, 106, 607 CrossRef CAS.
- I. Gomez-Pinto, E. Cubero, S. G. Kalko, V. Monaco, G. van der Marel, J. H. van Boom, M. Orozco and C. Gonzalez, J. Biol. Chem., 2004, 279, 24552 CrossRef CAS.
- K. Brown, B. E. Hingerty, E. A. Guenther, V. V. Krishnan, S. Broyde, K. W. Turteltaub and M. Cosman, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8507 CrossRef CAS.
- Y. Cai, D. J. Patel, N. E. Geacintov and S. Broyde, J. Mol. Biol., 2007, 374, 292 CrossRef CAS.
- F. A. Tanious, S. F. Yen and W. D. Wilson, Biochemistry, 1991, 30, 1813 CrossRef CAS.
- D. Sun, M. Hansen and L. Hurley, J. Am. Chem. Soc., 1995, 117, 2430 CrossRef CAS.
- H. Baruah and U. Bierbach, Nucleic Acids Res., 2003, 31, 4138 CrossRef CAS.
- Y. J. Chu, S. Sorey, D. W. Hoffman and B. L. Iverson, J. Am. Chem. Soc., 2007, 129, 1304 CrossRef CAS.
- Y. J. Chu, D. W. Hoffman and B. L. Iverson, J. Am. Chem. Soc., 2009, 131, 3499 CrossRef CAS.
- M. A. Nazif, J. A. Bangert, I. Ott, R. Gust, R. Stoll and W. S. Sheldrick, J. Inorg. Biochem., 2009, 103, 1405 CrossRef CAS.
- J. X. Dai, C. Punchihewa, P. Mistry, A. T. Ooi and D. Z. Yang, J. Biol. Chem., 2004, 279, 46096 CrossRef CAS.
- H. Baruah and U. Bierbach, J. Biol. Inorg. Chem., 2004, 9, 335 CrossRef CAS.
- R. Guddneppanavar, G. Saluta, G. L. Kucera and U. Bierbach, J. Med. Chem., 2006, 49, 3204 CrossRef CAS.
- H. Baruah, M. W. Wright and U. Bierbach, Biochemistry, 2005, 44, 6059 CrossRef CAS.
- S. Takenaka, Bull. Chem. Soc. Jpn., 2001, 74, 217 CrossRef CAS.
- W. A. Kibbe, Nucleic Acids Res., 2007, 35, W43 CrossRef.
- J. Vinje, J. A. Parkinson, P. J. Sadler, T. Brown and E. Sletten, Chem.–Eur. J., 2003, 9, 1620 CrossRef CAS; J. L. Beck, R. Gupta, T. Urathamakul., N. L. Williamson, M. M. Sheil, J. R. Aldrich-Wright and S. F. Ralph, Chem. Commun., 2003, 626 RSC; J. A. Parkinson, Y. Chen, P. D. Murdoch, Z. J. Guo, S. J. Berners-Price, T. Brown and P. J. Sadler, Chem.–Eur. J., 2000, 6, 3636 CrossRef CAS; D. R. Hare, D. Wemmer, S. H. Chou, G. Drobny and B. R. Reid, J. Mol. Biol., 1983, 171, 319 CrossRef CAS.
- S. L. Lam and S. C. F. Au-Yeung, J. Mol. Biol., 1997, 266, 745 CrossRef CAS.
- Y. Kwon, Z. Xi, L. S. Kappen, I. H. Goldberg and X. Gao, Biochemistry, 2003, 42, 1186 CrossRef CAS; G. Hwang, G. B. Jones and I. H. Goldberg, Biochemistry, 2004, 43, 641 CrossRef CAS.
- E. Alessio, Y. Xu, S. Cauci, G. Mestroni, F. Quadrifoglio, P. Viglino and L. G. Marzilli, J. Am. Chem. Soc., 1989, 111, 7068 CrossRef CAS.
- M. D. Reily and L. G. Marzilli, J. Am. Chem. Soc., 1986, 108, 8299 CrossRef CAS.
- V. M. Rodriguez-Bailey and M. J. Clarke, Inorg. Chem., 1997, 36, 1611 CrossRef CAS.
- A. Mukherjee, R. Lavery, B. Bagchi and J. T. Hynes, J. Am. Chem. Soc., 2008, 130, 9747 CrossRef CAS; M. J. L. Cocco, L. A. Hanakahi, M. D. Huber and N. Maizels, Nucleic Acids Res., 2003, 31, 2944 CrossRef CAS; Y. Coppel, J. F. Constant, C. Coulombeau, M. Demeunynck, J. Garcia and J. Lhomme, Biochemistry, 1997, 36, 4831 CrossRef CAS; W. D. Wilson, Y. Li and J. M. Veal, in Advances in DNA Sequence Specific Agents, ed. L. H. Hurley, Elsevier, New York 1992, p. 89 Search PubMed; J. Lee, V. Guelev, S. Sorey, D. W. Hoffman and B. L. Iverson, J. Am. Chem. Soc., 2004, 126, 14036 Search PubMed; X. G. Liang, H. Asanuma, H. Kashida, A. Takasu, T. Sakamoto, G. Kawai and M. Komiyama, J. Am. Chem. Soc., 2003, 125, 16408 CrossRef CAS.
- K. Wuthrich, NMR of Proteins and Nucleic Acids, John Wiley and Sons, Inc., USA, 1986, p. 30 Search PubMed.
- K. Kobayashi, Y. Asakawa, Y. Kikuchi, H. Toi and Y. Aoyama, J. Am. Chem. Soc., 1993, 115, 2648 CrossRef CAS; K. Gkionis, J. A. Platts and J. G. Hill, Inorg. Chem., 2008, 47, 3893 CrossRef CAS.
- J. Sola, A. Riera, X. Verdaguer and M. A. Maestro, J. Am. Chem. Soc., 2005, 127, 13629 CrossRef CAS; A. Uldry, J. M. Griffin, J. R. Yates, M. Pérez-Torralba, M. D. Santa Marıa, A. L. Webber, M. L. L. Beaumont, A. Samoson, R. M. Claramunt, C. J. Pickard and S. P. Brown, J. Am. Chem. Soc., 2008, 130, 945 CrossRef CAS.
- B. S. Thoma and K. M. Vasquez, Mol. Carcinog., 2003, 38, 1 CrossRef CAS.
- A. Luch, Nat. Rev. Cancer, 2005, 5, 113 CrossRef CAS; M. S. Greenblatt, W. P. Bennett, M. Hollstein and C. C. Harris, Cancer Res., 1994, 54, 4855 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Reactions of II with 15N-1. HPLC, HPLC-ESI-MS, NMR and pH measurements, Tables S1–S7 and Figs. S1–S7. See DOI: 10.1039/c0sc00175a |
| This journal is © The Royal Society of Chemistry 2010 |