Solvent-free catalytic enantioselective C–C bond forming reactions with very high catalyst turnover numbers†
Philip
Pelphrey
a,
Jørn
Hansen
b and
Huw M. L.
Davies
*b
aDepartment of Chemistry, University at Buffalo, The State University of New York, Buffalo, NY 14220, USA
bDepartment of Chemistry, Emory University, 1515 Dickey Drive, Atlanta, GA 30322, USA. E-mail: hmdavie@emory.edu; Fax: +1 404 727 7766; Tel: +1 404 727 6839
First published on 11th June 2010
Abstract
Rhodium(II)-catalyzed reactions of donor/acceptor carbenoids can be achieved with catalyst loadings as low as 6 × 10−7 mol equivalents when the reactions are conducted in the absence of solvent. ReactIR studies demonstrate that donor/acceptor carbenoids are ideal for achieving high catalyst turnover numbers due to the rapid reactions of their diazo precursors and the enhanced selectivity of this class of carbenoids.
A primary focus in the field of transition metal catalysis is the development of catalytic systems capable of achieving high turnover numbers (TONs).1 Low catalyst loadings increase the practicality of a process as many of the commonly used metal complexes are expensive and removal of the metal from the product is often a major challenge. Catalytic hydrogenation is the most established metal catalyzed asymmetric transformation for commercial scale reactions.2 Routinely, catalyst loadings of 0.1–0.01 mol%3 are employed, but specific examples are known in which the catalyst is capable of achieving TONs up to 2
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400000.4 Progress has been made to achieve high TONs in asymmetric C–C bond forming reactions,5–7 but they have not yet reached the same level of practicality as asymmetric hydrogenation.
400000.4 Progress has been made to achieve high TONs in asymmetric C–C bond forming reactions,5–7 but they have not yet reached the same level of practicality as asymmetric hydrogenation.
Practical high TON reactions require a relatively fast catalytic cycle and issues such as product inhibition and catalyst destruction need to be minimized. A promising strategy for achieving high TONs is to use high energy substrates that will facilitate a rapid catalytic cycle. Diazo compounds are substrates that meet this requirement because they can be decomposed by a variety of relatively stable metal catalysts to generate reactive metal carbenoids, with a driving force of forming N2 (Fig. 1).8 The subsequent reaction of the metal carbenoid to form the product and recovered catalyst is also very facile. In order to achieve high TONs, however, competing destructive reactions by the metal carbenoid need to be controlled.
 | ||
| Fig. 1 Diazo compounds as reagents for high TON catalytic cycles. | ||
In the case of the most common carbenoid precursor, ethyl diazoacetate, the typical catalyst loading is about 1 mol%, even though the reactions are generally very fast at ambient temperature,8,9 Lower catalyst loadings have been successful in certain reactions of ethyl diazoacetate, the most notable being the 11000 TONs reported for asymmetric cyclopropanation using a chiral ruthenium porphyrin catalyst.10 We have found that donor/acceptor-substituted carbenoids are much more selective than carbenoids with just an acceptor group,11,12 and can achieve much greater catalyst efficiency.13,14 However, in our previously developed procedure, highly enantioselective reactions with TONs approaching 100000 could be achieved only by using an exotic dirhodium catalyst Rh2(S-biTISP)2 with a bridged bis-carboxylate ligand (Fig. 2), an equivalent of methyl benzoate and large amounts of molecular sieves as additives. In this edge article, we demonstrate through ReactIR studies why donor/acceptor carbenoids are the optimum systems for low catalyst loadings. Furthermore, we describe a simple solvent-free method that can be applied to rhodium(II)-catalyzed carbenoid transformations with the much more standard catalysts, Rh2(S-DOSP)4 and Rh2(S-PTAD)4,15,16 resulting in TONs of >1000000.
 | ||
| Fig. 2 Dirhodium(II) catalysts. | ||
A central hypothesis behind this study is the idea that reactive carbenoids destroy the catalyst unless they are effectively trapped. Since donor/acceptor carbenoids are more stabilized than conventional carbenoids, their tendency to destroy the catalyst might be considerably diminished.11 In order to test this concept, the reactions of three types of diazo compounds with styrene in dichloromethane were monitored by ReactIR, tracking the disappearance of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N2 stretch frequencies. The reaction of methyl phenyldiazoacetate (1a) using Rh2(S-DOSP)415 as catalyst was very fast. When 0.1 mol% of catalyst was used, the reaction was complete in 3 s, while with 0.01 mol% of catalyst the reaction took 1 min to reach completion (Fig. 3). In contrast, the reaction of ethyl diazoacetate (2) with 0.1 mol% of Rh2(S-DOSP)4 initiated at a relatively fast rate, but the catalyst became inactive after about 400 TONs. This would explain why 1 mol% catalyst loading is typically used for the reactions with ethyl diazoacetate (2). The reaction with methyl diazomalonate (3) using 0.5 mol% of Rh2(S-DOSP)4 was slow, achieving less than 10% conversion in 3 min. These studies demonstrate that the more stabilized donor/acceptor carbenoids are best suited for achieving high TONs. They undergo very fast reactions while the catalyst remains active through many cycles.
N2 stretch frequencies. The reaction of methyl phenyldiazoacetate (1a) using Rh2(S-DOSP)415 as catalyst was very fast. When 0.1 mol% of catalyst was used, the reaction was complete in 3 s, while with 0.01 mol% of catalyst the reaction took 1 min to reach completion (Fig. 3). In contrast, the reaction of ethyl diazoacetate (2) with 0.1 mol% of Rh2(S-DOSP)4 initiated at a relatively fast rate, but the catalyst became inactive after about 400 TONs. This would explain why 1 mol% catalyst loading is typically used for the reactions with ethyl diazoacetate (2). The reaction with methyl diazomalonate (3) using 0.5 mol% of Rh2(S-DOSP)4 was slow, achieving less than 10% conversion in 3 min. These studies demonstrate that the more stabilized donor/acceptor carbenoids are best suited for achieving high TONs. They undergo very fast reactions while the catalyst remains active through many cycles.
 | ||
| Fig. 3 Effect of diazo structure on the relative rate of cyclopropanation using Rh2(S-DOSP)4 as catalyst (red line: 1a, S : C ratio 1000 : 1; blue line: 1a, S : C ratio 10000 : 1; green line: 2, S : C ratio 1000 : 1; grey line: 3, S : C ratio 200:1. | ||
The next series of experiments explored how this chemistry could be conducted with extremely low catalyst loadings. As a productive carbenoid reaction is bimolecular, whereas destruction of the catalyst via the carbenoid complex is likely to be unimolecular, it was reasoned that the highest TONs would be obtained in highly concentrated solutions. Consequently, solvent free conditions were explored for this chemistry. In a standard reaction, 50 mmol of the diazo compound and 50–100 mmol of the trapping agent were placed in a round-bottomed flask with an overhead stirrer. A specified amount of the catalyst was then added and the reaction monitored visually by nitrogen evolution and disappearance of the characteristic red color of the diazo compound. The catalysts used in this study were Rh2(S-DOSP)4 and Rh2(S-PTAD)4,16 two generally effective chiral catalysts for intermolecular reactions of donor/acceptor carbenoids.15,16
The first test was the reaction of 1a with styrene (eqn (1), Table 1).17 When using 0.01 mol% Rh2(S-DOSP)4, the reaction was too exothermic even at −20 °C. A better result was obtained using 0.003 mol% catalyst at −30 °C, as under these conditions the reaction went smoothly to completion in 3 h to generate the crude product in 75% ee. This material could be directly recrystallized from ethanol to generate 63% yield of the cyclopropane 4a in >99% ee.‡ As is typical for donor/acceptor carbenoids, the cyclopropanation reaction was highly diastereoselective (>95:5 d.r.).17,18 By decreasing the amount of Rh2(S-DOSP)4 to 0.0003 mol%, a controllable reaction could be achieved at ambient temperature in 5 days, resulting in the formation of 4a in 96% yield and 58% ee. Rh2(S-PTAD)4 appears to be even more active and a complete reaction could be achieved in 4 days with just 0.00005 mol% catalyst, though with significantly diminished enantiomeric excess (13% ee). The reaction with methyl p-(methoxy)phenyldiazoacetate (1b), which generates a more stabilized carbenoid, gave further improvement as only 0.00005 mol% of Rh2(S-PTAD)4 was sufficient to form 4b in 92% yield and 51% ee within 3 days (1.8 × 106 TONs).
 | (1) |
| Entry | Ar | Catalyst | Catalyst loading (mol%) | Temp./°C | Time/h | Yield (%) | ee (%) | TON | TOF/h−1 |
|---|---|---|---|---|---|---|---|---|---|
| a After recrystallization. b Opposite enantiomer of drawn product formed. | |||||||||
| 1 | Ph | Rh2(S-DOSP)4 | 0.003 | −30 | 3 | 63a | 75 (99a) | 2.0 × 104 | 6.6 × 103 |
| 2 | Ph | Rh2(S-DOSP)4 | 0.0003 | 25 | 120 | 96 | 58 | 3.0 × 105 | 2.5 × 103 |
| 3 | Ph | Rh2(S-PTAD)4 | 0.00005 | 25 | 96 | 93 | 13b | 1.5 × 106 | 1.5 × 104 |
| 4 | p-(MeO)Ph | Rh2(S-DOSP)4 | 0.0001 | 25 | 144 | 90 | 69 | 8.5 × 105 | 5.9 × 103 |
| 5 | p-(MeO)Ph | Rh2(S-PTAD)4 | 0.00005 | 25 | 72 | 92 | 51b | 1.8 × 106 | 2.5 × 104 |
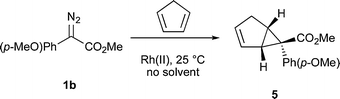
The cyclopropanation of cyclopentadiene could also be achieved with very high TONs (eqn (2), Table 2). The Rh2(S-DOSP)4 catalyzed reaction of 1b using 0.0003 mol% of catalyst gave cyclopropane 5 in 83% yield and 78% ee. The Rh2(S-PTAD)4 catalyzed reaction was more effective and 0.00006 mol% was sufficient to form 5 in 83% yield and 76% ee.
 | (2) |
Since the solvent-free reaction conditions are relatively exothermic, care is required for large-scale reactions. The reactions are best started with a minute amount of catalyst, followed by addition of more catalyst as needed. Purification is simply achieved by distillation or recrystallization. These reaction conditions are very different from what would typically be used with the more conventional carbenoid derived from ethyl diazoacetate, in which the diazo compound would be added slowly by syringe pump, in order to avoid carbene dimerization.
Styryldiazoacetates are another class of diazo compounds that give donor/acceptor carbenoid intermediates. The Rh2(S-DOSP)4 catalyzed reaction of 6 with styrene using 0.003 mol% catalyst generated the valuable chiral building block 7 (eqn (3))15,19 in 72% yield and 81% ee. One recrystallization enriched the material to >99% ee. The reaction of 6 with cyclopentadiene, catalyzed by 0.003 mol% of Rh2(S-DOSP)4, generated the [4 + 3] cycloadduct 8 in 84% yield and 81% ee (eqn (4)).20
 | (3) |
 | (4) |
The donor/acceptor carbenoids are very effective at intermolecular C–H functionalization by direct insertion into C–H bonds.21 These reactions can be carried out with relatively high TONs. 1,4-Cyclohexadiene is very reactive towards C–H insertion and reaction of 1a using 0.01 mol% Rh2(S-DOSP)4 at 0 °C generated 9 in 96% yield and 81% ee (eqn (5)).
 | (5) |
In summary, these studies demonstrate that reactions involving donor/acceptor substituted rhodium carbenoids can be conducted with very high TONs in the absence of solvent. When very active trapping agents are used, such as styrene and cyclopentadiene, the catalyst to substrate ratio can be as low as 0.6 ppm, resulting in TONs of up to 1.8 million. Donor/acceptor carbenoids are ideal for reactions with low catalyst loadings because the carbenoids are more stabilized than other classes of carbenoids. They are still very reactive, but less prone to catalyst destruction and carbene dimerization. The solvent-free conditions greatly enhance the efficiency of the catalysts, presumably by favouring the productive bimolecular process for product formation over the unimolecular process for metal carbenoid destruction.
This material is based upon work supported by the National Science Foundation under the Center for Chemical Innovation in Stereoselective C–H Functionalization (CHE-0943980) and CHE-0750273.
Notes and references
- J.-C. Hierso, M. Beaupérin and P. Meunier, Eur. J. Inorg. Chem., 2007, 3767 CrossRef CAS
, and references therein..
-
Asymmetric Catalysis on Industrial Scale, ed. H. U. Blaser and E. Schmidt, Wiley-VCH, Weinheim, 2004 Search PubMed
- W. S. Knowles and R. Noyori, Acc. Chem. Res., 2007, 40, 1238 CrossRef CAS
- H. Doucet, T. Ohkuma, K. Murata, T. Yokozawa, M. Kozawa, E. Katayama, A. F. England, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed., 1998, 37, 1703 CrossRef CAS
- A. T. Axtell, C. J. Cobley, J. Klosin, G. T. Whiteker, A. Zanotti Gerosa and K. A. Abboud, Angew. Chem., Int. Ed., 2005, 44, 5834 CrossRef CAS
- K. Mikami, Y. Kawakami, K. Akiyama and K. Aikawa, J. Am. Chem. Soc., 2007, 129, 12950 CrossRef CAS
- M. P. Doyle, I. M. Phillips and W. H. Hu, J. Am. Chem. Soc., 2001, 123, 5366 CrossRef CAS
-
M. P. Doyle, M. A. McKervey, T. Ye, Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides, John Wiley & Sons, Inc., New York, 1998 Search PubMed
- M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704 CrossRef CAS
- C.-M. Che, J.-S. Huang, F.-W. Lee, Y. Li, T.-S. Lai, H.-L. Kwong, P.-F. Teng, W.-S. Lee, W.-C. Lo, S.-M. Peng and Z.-Y. Zhou, J. Am. Chem. Soc., 2001, 123, 4119 CrossRef CAS
- J. Hansen, J. Autschbach and H. M. L. Davies, J. Org. Chem., 2009, 74, 6555 CrossRef CAS
- H. M. L. Davies and S. A. Panaro, Tetrahedron, 2000, 56, 4871 CrossRef CAS
- H. M. L. Davies and C. Venkataramani, Org. Lett., 2003, 5, 1403 CrossRef CAS
- J. R. Manning and H. M. L. Davies, Org. Synth., 2007, 84, 334 CAS
- H. M. L. Davies, P. R. Bruzinski, D. H. Lake, N. Kong and M. J. Fall, J. Am. Chem. Soc., 1996, 118, 6897 CrossRef CAS
- R. P. Reddy, G. H. Lee and H. M. L. Davies, Org. Lett., 2006, 8, 3437–3440 CrossRef CAS
- H. M. L. Davies, P. R. Bruzinski and M. J. Fall, Tetrahedron Lett., 1996, 37, 4133 CrossRef CAS
- D. T. Nowlan, T. M. Gregg, H. M. L. Davies and D. A. Singleton, J. Am. Chem. Soc., 2003, 125, 15902 CrossRef CAS
- E. J. Corey and T. G. Gant, Tetrahedron Lett., 1994, 35, 5373 CrossRef CAS
- H. M. L. Davies, D. G. Stafford, B. D. Doan and J. H. Houser, J. Am. Chem. Soc., 1998, 120, 3326 CrossRef CAS
- H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Representative procedures and spectral data. See DOI: 10.1039/c0sc00109k |
| ‡ Representative experimental procedure (Table 1, entry 1): methyl phenyldiazoacetate 1a (8.81 g, 50 mmol) was mixed with styrene (10 mL, 87 mmol, 1.7 equiv.) and cooled to −30 °C under an Ar atmosphere. Rh2(S-DOSP)4 (3.0 mg, 0.0016 mmol) was then added and the reaction allowed to stir for 3 h at −30 °C. Excess trapping agent was then removed by Kügelrohr distillation to give 4a (11.5 g, 91%, 75% ee). The material was recrystallized from ethanol to afford 4a as a white solid (8.0 g, 63%, >99% ee). Safety note: since the solvent-free reaction conditions are relatively exothermic, care is required for conducting reactions beyond the 50 mmol scale reported herein. The reactions are best started with a minute amount of catalyst, followed by addition of more catalyst as needed. Due to the potential instability of diazo compounds, excessive heating should be avoided and the work should be conducted behind a blast shield. |
| This journal is © The Royal Society of Chemistry 2010 |