Comparison and improvement of the existing methods for the determination of aflatoxins in human serum by LC-MS/MS
Antonello
Santini
*a,
Rosalia
Ferracane
a,
Giuseppe
Meca
b and
Alberto
Ritieni
a
aDepartment of Food Science, University of Naples “Federico II”, Via Università, 100 - 80055, Portici, Naples, Italy. E-mail: asantini@unina.it.; Fax: +0039 81 2539317; Tel: +0039 81 2539317
bDepartamento de Medicina Preventiva, Universidad de Valencia, Avenida Vicent Andres Estelles, 46100, Burjassot, Valencia, Spain
First published on 20th May 2010
Abstract
Aflatoxins are toxic metabolites of some Aspergillus flavus, A. parasiticus and A. nomius strains that occur in many foods and feeds. Aflatoxin B1 is the most abundant and toxic member of the family, and it is also the most potent hepatocarcinogen known. In order to estimate the potential human health risk of aflatoxin B1, it is useful to measure its blood concentration. In this work a rapid liquid chromatography coupled to mass spectrometry (LC-MS/MS) method for the simultaneous determination of aflatoxins B1, B2, G1 and G2 and its metabolites, aflatoxins M1 and M2 in human serum, is proposed. Small serum volumes have been used and the extracts have been obtained without any clean-up procedure. The isolation of the aflatoxins from human serum was achieved using subsequent pH-controlled solvent extractions steps. Recovery rates in the range between 31 and 98% were achieved in the serum, while the limits of detection and quantification for the proposed method were 0.5 ng mL−1 and 1.0 ng mL−1, respectively. The validation of the method has been demonstrated by analysing spiked and naturally contaminated blood serum.
Introduction
Aflatoxins are secondary metabolites produced by Aspergillus flavus, A. parasiticus and A. nomius, and they are named aflatoxins B1, B2, G1 and G2. Fig. 1 shows the chemical structure of aflatoxins, that incorporates dihydrofuran and tetrahydrofuran moieties coupled to a substituted coumarin. Aflatoxins can be converted to more reactive, electrophilic epoxides by phase I metabolism occurring primarily in the liver, and have been frequently detected in samples of corn, peanuts, cottonseeds, rice and other foodstuffs.1,2,3 Among these toxins, the aflatoxin B1 is the most studied, the most commonly occurring one4 and the one with the highest mutagenic and carcinogenic activity.5 Several epidemiological studies have found positive association between aflatoxin B1 (AFB1) dietary exposure and an increased risk of human liver cancer.6–9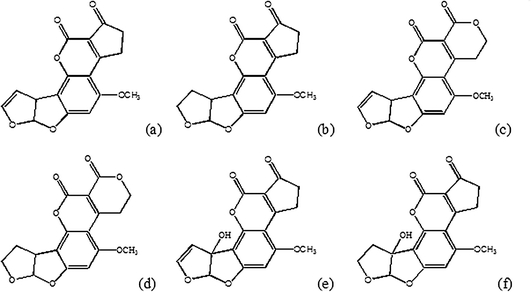 | ||
| Fig. 1 Chemical structures of aflatoxins B1 (a), B2 (b), G1 (c), G2 (d) and M1 (e), M2 (f). | ||
Cytochrome P-450 enzymes further convert aflatoxins to different metabolites,10e.g. aflatoxin B1 is converted to metabolites like aflatoxin B1-epoxide and the hydroxylated aflatoxins M1, P1 and Q1, as shown in Fig. 2. The hydroxylated metabolites form glucuronide and sulfate conjugates that can be enzymatically hydrolysed by β-glucuronidase and sulfatase.11
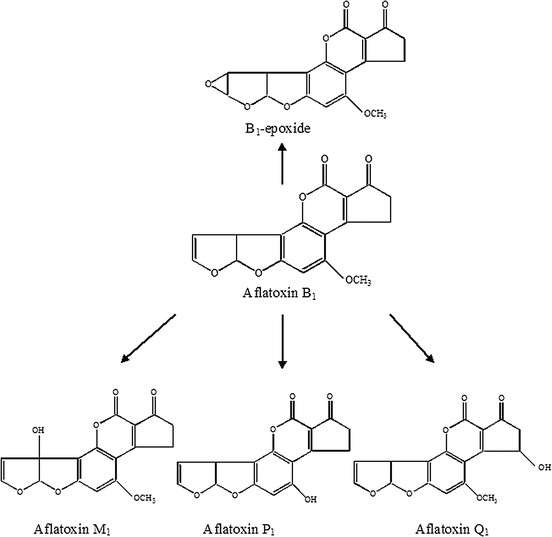 | ||
| Fig. 2 Metabolic pathways of aflatoxins B1 by cytochrome P-450 enzymes. | ||
Different analytical methods have been reported in the literature in order to facilitate the investigation of the role of ingested aflatoxins in small volumes of human sera.12,13 AFB1 has been extracted from 1 mL or less of human sera spiked with a known concentration of aflatoxin B1 and analyzed using high-performance liquid chromatography (HPLC) as the detection system. Several methods have been used to analyze feeds, foods and body fluids (human and animal plasma, serum, milk, etc.)14–18 The ELISA7 or radioimmunoassay (RIA) methods19,20 allow the quantification of the total amount of aflatoxins and results are expressed in term of aflatoxin B1 equivalents. Both methods however involve the use of specific antibodies not commercially available. The International Agency for Research on Cancer has classified AFB1 as a human carcinogen and aflatoxins B2, G1 and G2 (AFB2, AFG1 and AFG2) as possible nephrotoxic natural compounds and carcinogenic to humans.21,22 Due to carryover in food and feed they are considered nowadays to have the most severe impact of all mycotoxins on human health. Maximum residue levels have been set down to the ppt range in a wide variety of agricultural commodities, food, feed and milk, e.g. 0.01 μg kg−1 of AFM1 in milk for infants.23 Methods like LC/MS have been repeatedly used for structural elucidation in metabolism on aflatoxin containing analytes and specific matrices but only a limited number of quantitative methods have been published to determine the more common aflatoxins present in food,24–28,35–38 milk,27 cheese,39 herbs,40 urine,31–34,41 airborne dust41 and cigarette smoke.42 LC/MS has been used as a confirmation technique for the already well established, reliable and robust LC-FL methodology29,30,36,41 and has also been used to confirm positive results of TLC and ELISA based screening analyses. All the aflatoxins exhibit good ESI ionisation efficiency in the positive ion mode with abundant protonated molecules [MH]+ and sodium adduct ions36,40,41 and typically, for AFB1, AFB2, AFG1 and AFG2, the formation of sodium adduct ions can easily be suppressed by the addition of ammonium ions to the mobile phase leading to a better MS sensitivity.39
Reports about the utility of APCI interfaces and ionization efficiencies in this mode seem to be highly dependent on the aflatoxin studied and the APCI interface geometry.29,30 A major focus of this work has been the development of a rapid extraction method of the non protein conjugated aflatoxins, without any cleanup procedure, from human sera and the use of LC-MS/MS technique for their analysis and quantification.
This method has been proved to be more sensitive for the simultaneous determination of aflatoxins B1, B2, G1, G2, M1, M2, and moreover smaller sample volumes of serum can be used for the analysis. Aflatoxins are in free equilibrium with the albumin combined form and it is reported in the literature the effect of pH and/or serum concentration of fatty acids on the formation of the adducts.43 Moreover, a recent study showed that green tea polyphenols might modulate the formation of the adducts between aflatoxin B1 and albumin.44
Advanced spectrometric methods, such as LC-MS/MS, permit quantification and recognition of the free aflatoxins in the sera with fewer problems on recovery, sensitivity and chemical identification.1, 18 All these reasons support the development of a new method to analyze sera with the aim of evaluating the aflatoxin exposure directly from their free forms.
Experimental
Chemicals and reagents
Blood specimens were obtained, by antecubital venipuncture, from healthy adult volunteers. After storing the blood for 2 h, serum was separated by centrifugation.Aflatoxins B1, B2, G1, G2, M1 and M2 were purchased from Sigma-Aldrich (Milano, Italy) and stored at 4 °C. Standard stock solutions were prepared by dissolving standards in methanol to give a concentration of 1mg mL−1 for each solution.
Water, acetonitrile and methanol for LC mobile phase were HPLC-grade (Merck, Darmstadt, Germany). Formic acid was obtained from Fluka (Milano, Italy). The organic solvents used for the extraction as described in the following were all purchased from Carlo Erba (Milano, Italy) and the water used was purified with a Milli-Q system (Millipore, MA, USA).
Instrumentation
Mobile phase A consisted of H2O containing 0.2% formic acid, while mobile phase B consisted of CH3OH–CH3CN mixture (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 v/v). The following binary gradient was applied: initial condition 20% B; 0–12 min, 80% B; 12–13 min, 100% B; 13–18 min constant at 100% B; finally returning to the initial conditions in 2 min.
:10 v/v). The following binary gradient was applied: initial condition 20% B; 0–12 min, 80% B; 12–13 min, 100% B; 13–18 min constant at 100% B; finally returning to the initial conditions in 2 min.
The acquisition was carried out in MRM (Multiple Reaction Monitoring) in positive ion mode for each compounds. Data acquisition and processing were performed using Analyst software v. 1.4. Acquisition parameters are summarised in the Table 1.
| Analyte | Retention time/min | Precursor Ion | Product Ions | DP | CE |
|---|---|---|---|---|---|
| Aflatoxin B1 | 11.3 | 313.1 | 269 | 42 | 42 |
| [M + H]+ | 285 | 36 | |||
| Aflatoxin B2 | 10.9 | 315 | 259 | 56 | 39 |
| [M + H]+ | 287 | 38 | |||
| Aflatoxin G1 | 10.4 | 329 | 243 | 58 | 39 |
| [M + H]+ | 283 | 37 | |||
| Aflatoxin G2 | 9.94 | 331 | 313 | 55 | 36 |
| [M + H]+ | 245 | 43 | |||
| 257 | 44 | ||||
| Aflatoxin M1 | 9.93 | 329 | 273 | 50 | 33 |
| [M + H]+ | 259 | 32 | |||
| 301 | 26 | ||||
| Aflatoxin M2 | 9.28 | 331 | 273 | 50 | 30 |
| [M + H]+ | 285 | 31 | |||
| 259 | 31 | ||||
:30, v/v) and used for LC/MS/MS analysis.
In the second method according to Nelson et al.,46 500 μl of serum samples in duplicate were extracted using chloroform and hexane. Initially, 1mL hexane was added to 1 mL aliquot of aflatoxin spiked serum and mixed for 2 min, the mixture was centrifuged for 5 min at 3000 rpm and the upper hexane layer containing the serum lipids, removed. This procedure was repeated twice and the hexane layer was removed after each centrifugation. The serum was then extracted four times with 1 mL aliquots of chloroform; after vigorous shaking for 4 min, samples were centrifuged for 10 min at 3000 rpm and the lower chloroform layer removed. The chloroform extracts were pooled, and evaporated using a centrifugal evaporator (Savant Instruments Inc., Farmingdale, NY, USA). At the end of the procedure, the dry extracts were dissolved in 200 μL of a solution of CH3OH–H2O (70:30, v/v) before the analysis by LC-MS/MS.
The third procedure of extraction, here proposed, follows the method proposed in year 1991 by Beker et al.46 for the ochratoxin extraction from human serum with slight modifications. In particular: serum samples (500μL) were added to 2.5 mL ethyl acetate and mixed. The mixture was centrifuged for 10 min at 1500 rpm and the over layer recovered. This procedure was repeated twice and then the residue was evaporated using a centrifugal evaporator (Savant Instruments Inc., Farmingdale, NY, USA) and dissolved in 500 μL mixture of CH3OH–H2O (70:30, v/v).
In the fourth procedure, validated, samples were extracted as described for the third method, with the difference that ethyl acetate was added to each serum sample and samples were acidified to obtain a final pH value equal to 2. The samples were then extracted, dried and dissolved in 500 μL of a mixture CH3OH–H2O (70:30, v/v), by agitating vigorously and finally filtered on RC 0.2 μm Syringe Filters (Phenomenex, Italy) before the LC-MS/MS analysis.
:30, v/v). Samples were spiked at 20 μg mL−1 and samples were prepared for the analysis using the three different methods as described above.
To validate the method without cleanup procedures, three levels of contamination: 5, 10 and 20 μg L−1 were tested. The samples were left overnight at room temperature and then were extracted and analyzed. Three replicates were analyzed for each spiked serum extract.
The precision of the method in terms of repeatability (intra-day precision) and reproducibility (inter-day precision) was evaluated by calculating the relative standard deviation (RSD) as reported respectively in Table 2(A) and (B). The RSDs of the intra-day were in the range of 3.5–8.9%. while the RSDs of the inter-day were in the range of 3.8–9.1%. Table 3 reports the estimated parameters of the regression lines (y = a + bx) for the calibration curves.
| (A) | ||||||
|---|---|---|---|---|---|---|
| Aflatoxin | Contamination level 20 μg L−1 | RSD (%) | Contamination level 10 μg L−1 | RSD (%) | Contamination level 5 μg L−1 | RSD (%) |
| AF B1 | 82.0 ± 6.2 | 7.6 | 80.8 ± 6.7 | 8.3 | 79.1 ± 5.1 | 6.4 |
| AF B2 | 98.5 ± 7.2 | 7.3 | 97.8 ± 7.4 | 7.6 | 96.1 ± 5.8 | 6.0 |
| AF G1 | 31.4 ± 2.5 | 8.0 | 31.8 ± 1.1 | 3.5 | 30.9 ± 1.7 | 5.5 |
| AF G2 | 62.5 ± 4.2 | 6.7 | 61.5 ± 5.3 | 8.6 | 60.5 ± 3.8 | 6.3 |
| AF M1 | 70.5 ± 5.6 | 7.9 | 68.8 ± 5.1 | 7.4 | 67.7 ± 4.1 | 6.1 |
| AF M2 | 73.5 ± 5.7 | 7.8 | 70.8 ± 6.3 | 8.9 | 69.5 ± 5.2 | 7.5 |
| (B) | ||||||
| Aflatoxin | Contamination level 20 μg L−1 | RSD (%) | Contamination level 10 μg L−1 | RSD (%) | Contamination level 5 μg L−1 | RSD (%) |
| AF B1 | 83.6 ± 5.5 | 6.6 | 81.6 ± 5.6 | 6.9 | 77.8 ± 7.1 | 9.1 |
| AF B2 | 97.1 ± 6.7 | 6.9 | 96.8 ± 3.7 | 3.8 | 97.1 ± 8.6 | 8.9 |
| AF G1 | 33.4 ± 2.1 | 6.3 | 32.3 ± 1.8 | 5.6 | 32.1 ± 1.9 | 5.9 |
| AF G2 | 63.6 ± 4.6 | 7.2 | 62.5 ± 5.3 | 8.5 | 61.8 ± 4.8 | 7.8 |
| AF M1 | 70.9 ± 4.8 | 6.8 | 69.6 ± 5.7 | 8.0 | 68.5 ± 4.2 | 6.1 |
| AF M2 | 73.9 ± 6.5 | 8.8 | 72.5 ± 5.4 | 7.4 | 70.2 ± 5.9 | 8.4 |
| Aflatoxin | Slope (b) | Intercept (a) | R2 |
|---|---|---|---|
| AF B1 | 1800 | −624 | 0.998 |
| AF B2 | 561 | 1140 | 0.996 |
| AF G1 | 872 | 4200 | 0.995 |
| AF G2 | 595 | 320 | 0.996 |
| AF M1 | 711 | 2160 | 0.995 |
| AF M2 | 391 | 1740 | 0.996 |
:30, v/v).
The peak area of each toxin was plotted against the concentration, and the calibration curves were calculated using the linear regression method. For the determination of the detection limit (LOD), the signal to noise (S/N) ratio was calculated (peak to peak analysis with Analyst software v. 1.4) from the calibration curve for each toxin, and the LOD was determined by S/N to be more than 3:1. The limit of quantification (LOQ) was obtained by S/N to be more than 10:1.
Results and discussion
Development of sample preparation
The multiple extraction procedure was optimized after comparing the performance of four different methodologies for the aflatoxins analysis in physiological samples. The validation was obtained by comparing the recoveries of analyte-free sample, for each method, by spiking at level of 20 μg L−1. The results for all mycotoxins analyzed are reported in Table 4.| Aflatoxin | Method I | RSD% | Method II | RSD% | Method III | RSD% | Method IV | RSD% |
|---|---|---|---|---|---|---|---|---|
| % recovery | % recovery | % recovery | % recovery | |||||
| AF B1 | 2.0 ± 0.17 | 8.5 | 87 ± 5.9 | 6.8 | 35.4 ± 3.1 | 8.8 | 82.0 ± 6.2 | 7.6 |
| AF B2 | 4.2 ± 0.38 | 9.0 | 40.5 ± 3.6 | 8.9 | 31.0 ± 2.6 | 8.4 | 98.5 ± 7.2 | 7.3 |
| AF G1 | 6.8 ± 0.65 | 9.6 | 58.0 ± 4.1 | 7.1 | 30.5 ± 2.7 | 8.9 | 31.4 ± 2.5 | 8.0 |
| AF G2 | 7.3 ± 0.63 | 8.6 | 38.0 ± 3.3 | 8.7 | 47.4 ± 4.1 | 8.6 | 62.5 ± 4.2 | 6.7 |
| AF M1 | 9.2 ± 0.79 | 8.6 | 61.0 ± 3.7 | 6.1 | 47.3 ± 4.2 | 8.9 | 70.5 ± 5.6 | 7.9 |
| AF M2 | 6.0 ± 0.59 | 9.0 | 32.0 ± 2.8 | 8.8 | 58.3 ± 4.8 | 8.2 | 73.5 ± 5.7 | 7.8 |
Using the method proposed by Shephard et al.45 the percentage recovery ranged from 2% to 9.2%. In the procedure described by Nelson et al.,46 the percentage recovery, ranged from 32% and 87%, and was higher than the one obtained using the third protocol of extraction, proposed by Beker et al.47 ranged from 30.5% to 58.3%. The novel method proposed here for the simultaneous quantitative extraction from serum, which consisted of acidifying the extraction mixture, was revealed to be the most appropriate due to the structural characteristics of the aflatoxins. The obtained recovery values are in the range 31.4% to 91.5%, suggesting that this last procedure is the most appropriate one.
In this study the main goal was to identify a rapid and simple method for the extraction of aflatoxins by comparing different techniques and evaluating the influence of pH during this phase. For this reason, four recovery experiments were carried out to a spiking level of 20 μg L−1 for each extraction method. In the first experiment, as described by Shepard et al.45 for fumonisins, samples were extracted with acetonitrile, for analogy. In the second experiment serum samples were extracted as described by Nelson et al.46 and dried; the third experiment used the procedure described by Beker et al.47 for ochratoxins and ethyl acetate was added to the serum samples during the extraction. In the fourth experiment serum samples were acidified to pH 2 and dried after centrifugation. All the samples were extracted and analyzed in triplicate and the reported results averaged. Accuracy and precision of the validated method were obtained by recovery experiments. Three different levels of contamination, namely 20 ppb, 10 ppb, 5 ppb, respectively, were confirmed for spiked human serum. The results for all the analyzed mycotoxins are reported in Table 2(A) and (B) where intra-day and inter-day precision of the LC/MS-MS method is shown.
LC/MS optimization and performance validation
Calibration curves for all the analytes are linear over the working range of 1–50 μg L−1. This is probably due to the very high response of the studied compounds. Squared correlation coefficients (R2) were in the range 0.995–0.998 for all point calibration curves, depending on the mycotoxin analyzed. Method detection limit (LOD) was estimated at the 500 ng L−1 level and quantification value (LOQ) was 1 μg L−1 for each analyzed sample solution and for the aflatoxin standards.An LC-ESI-MS/MS total ion chromatogram (TIC) of a blank serum sample spiked with six aflatoxins at level of 20 μg L−1 is shown in Fig. 3.
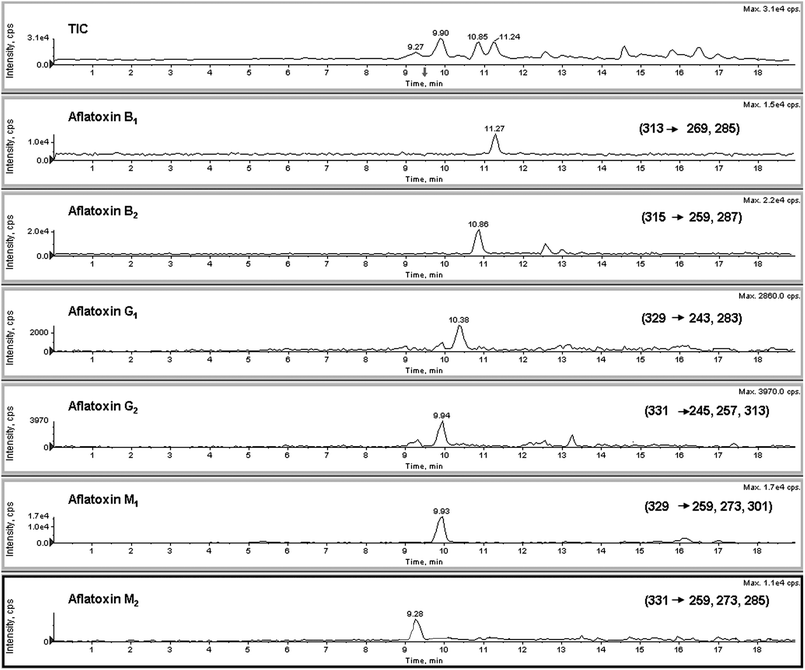 | ||
| Fig. 3 LC-ESI-MS/MS total ion chromatogram (TIC) of a blank serum sample spiked with six aflatoxins at level of 20 μg L−1. | ||
Conclusions
The method proposed, involves acidifying and analyzing dry blood serum samples after centrifugation, and presents several advantages with respect to other described methods for determining different mycotoxins in human serum; in particular it provides more rapid sample preparation without any clean-up steps, and uses a multi components analysis with a single chromatographic run. In addition, the in-house performance in terms of accuracy (recovery) and precision (repeatability) are quite satisfactory and superior with respect to the other published methods (ranged from 31.4% to 98.5%). Determination by LC-MS/MS of non-conjugated aflatoxins in serum was suggested and has been optimized as described in this paper because non-conjugated aflatoxins are most likely to cause biological effects or damage to tissues and organs through the circulatory system.Further studies are in progress in our laboratories to confirm the relationship that exists between serum aflatoxin levels and aflatoxins intake through the diet, and from this aspect the proposed method could be successfully applied to assess human exposure to numerous different aflatoxins. Moreover, the proposed method could be effectively used as a rapid and non-invasive tool to identify extremely advanced liver diseases in humans, which may be attributed to the ingestion of aflatoxins from foodstuff.
Acknowledgements
The authors wish to thank Dr. Maria Carmela Somma for her technical help and assistance and the PhD program “Cinc Segles” of the University of Valencia, Spain.References
- B. Huang, H. Zheng, C. Zengxuan, W. Yongjiang and R. Yiping, Anal. Chim. Acta, 2010, 662, 62 CrossRef CAS.
- I. Ruston, Food Chem., 1997, 59, 57 CrossRef.
- F. Galvano, V. Galofaro and G. Galvano, J. Food Prot., 1996, 59, 1070.
- P. Simon, P. Dlsaut, M. Lafontaine, Y. Morele and T. Nicot, J. Chromatogr., B, 1998, 712, 95 CrossRef CAS.
- I. Dvorackova, Aflatoxins and Human Health, CRC Press, Boca Raton, FL, 1990 Search PubMed.
- D. L. Sudakin, J. Toxicol., Toxin Rev., 2003, 23, 195 Search PubMed.
- J. Q. Zhu, L. S. Zhang, X. Hu, Y. Xiao, J. S. Chen, Y. C. Xu, J. Fremy and F. S. Chu, Cancer Res., 1987, 47, 1848 CAS.
- J. D. Groopman and K. F. Donahue, J. Assoc. Off. Anal. Chem., 1988, 71, 861 CAS.
- T. A. Bean and D. M. Yourtee, J. Toxicol. Toxin Rev., 1989, 8, 43 Search PubMed.
- D. L. Eaton, H. S. Ramsdell and G. E. Neal, in D. L. Eaton and J. D. Groopman (Editors), The Toxicology of Aflatoxins, Academic Press, San Diego, CA, 1994, p.45 Search PubMed.
- C. I. Wei, M. R. Marshall and D. P. H. Hsieh, Food Chem. Toxicol., 1985, 23, 809 CrossRef CAS.
- L. Grio, S. Jose, G. Frenich, A. Martinez Vidal, J. Luis and R. Romero-Gonzalez, J. Sep. Sci., 2010, 33, 502 CrossRef.
- F. Yuanjing, Y. Yi, X. Huiming, P. Bingnan, G. Haicheng, M. Fanli, Y. Miaomiao, C. Wei and H. Wendong, Faming Zhuanli Shenqing Gongkai Shuomingshu, 2010, 1, 1–9 Search PubMed.
- S. Hodisan, M. Sebesan, A. Cozma, Alina and A. Caraban, Fascicula Chimie, 2009, 16, 152 Search PubMed.
- L. Rampone, A. L. Piccinelli, L. Aliberti and L. Rastrelli, J. Sep. Sci., 2009, 32, 3837 CrossRef.
- S. Monbaliu, C. Van Poucke, C. Detavernier, F. Dumoulin, M. Van De Velde, E. Schoeters, S. Van Dyck, O. Averkieva, C. Van Peteghem and S. De Saeger, J. Agric. Food Chem., 2010, 58, 66 CrossRef CAS.
- H. Takeuchi, H. Isshiki, K. Hayashi, H. Kawai, Y. Hayashizaki, Y. Ohgaki and K. Shimura, Mie-ken Hoken Kankyo Kenkyusho Nenpo, 2009, 11, 29 Search PubMed.
- A. Santini, R. Ferracane, G. Meca and A. Ritieni, Anal. Bioanal. Chem., 2009, 395, 1253 CrossRef CAS.
- J. Tang and P. Guangchang, Shipin Kexue, 2009, 30, 326 Search PubMed.
- P. Li, Q. Zhang and W. Zhang, TrAC, Trends Anal. Chem., 2009, 28, 1115 CrossRef CAS.
- IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, International Agency for Research on Cancer, Lyon, 1993, p. 489 Search PubMed.
- Commission Regulation (EC) No. 1525/98, amending Regulation (EC) No 184/97 setting maximum residue levels for certain contaminants in foodstuffs, Official J. Eur. Commun. L 201/43, 1998, 43.
- J. D. Groopman, P. R. Donahue, J. Zhu, J. Chen and G. N. Wogan, Biochemistry, 1985, 82, 6492 CAS.
- E. Papp, K. H. Otta, G. Zaray and E. Mincsovics, Microchem. J., 2002, 73, 39 CrossRef CAS.
- S. Biselli, L. Hartig, H. Wegener and C. Hummert, LC–GC Eur., 2004, 17, 25 CAS.
- S. Biselli, L. Hartig, H. Wegener and C. Hummert, Spectroscopy, 2005, 20, 20 Search PubMed.
- L. K. Sorensen and T. H. Elbaek, J. Chromatogr., B, 2005, 820, 183 CrossRef CAS.
- M. Kokkonen, M. Jestoi and A. Rizzo, Food Addit. Contam., 2005, 22, 449 CrossRef CAS.
- H. K. Abbas, W. P. Williams, G. L. Windham, H. C. Pringle, W. Xie and W. T. Shier, J. Agric. Food Chem., 2002, 50, 5246 CrossRef CAS.
- H. K. Abbas, R. D. Cartwright, W. Xie and W. T. Shier, Crop Prot., 2006, 25, 1 CrossRef CAS.
- M. Walton, P. Egner, P. F. Scholl, J. Walker, T. W. Kensler and J. D. Groopman, Chem. Res. Toxicol., 2001, 14, 919 CrossRef CAS.
- P. A. Egner, X. Yu, J. K. Johnson, C. K. Nathasingh, J. D. Groopman, T. W. Kensler and B. D. Roebuck, Chem. Res. Toxicol., 2003, 16, 1174 CrossRef CAS.
- J. L. Wang-Buhler, S. J. Lee, W. G. Chung, J. F. Stevens, H. P. Tseng, T. H. Hseu, C. H. Hu, M. Westerfield, Y. H. Yang, C. L. Miranda and D. R. Buhler, Comp. Biochem. Physiol., Part C Toxicol. and Pharmacol., 2005, 140, 209 Search PubMed.
- P. F. Scholl, S. M. Musser and J. D. Groopman, Chem. Res. Toxicol., 1997, 10, 1144 CrossRef CAS.
- M. Vahl and K. Jorgensen, Z. Lebensm.-Unters. -Forsch. A, 1998, 206, 243 CrossRef CAS.
- J. Blesa, J. M. Soriano, J. C. Molto, R. Marin and J. Manes, J. Chromatogr., A, 2003, 1011, 49 CrossRef CAS.
- T. F. Schatzki and W. F. Haddon, J. Agric. Food Chem., 2002, 50, 3062 CrossRef CAS.
- M. Takino, T. Tanaka, K. Yamaguchi and T. Nakahara, Food Addit. Contam., 2004, 21, 76 CrossRef CAS.
- C. Cavaliere, P. Foglia, E. Pastorini, R. Samperi and A. Lagana, J. Chromatogr., A, 2006, 1101, 69 CrossRef CAS.
- M. Ventura, A. Gomez, I. Anaya, J. Diaz, F. Broto, M. Agut and L. Comellas, J. Chromatogr., A, 2004, 1048, 25 CrossRef.
- A. Kussak, C. A. Nilsson, B. Andersson and J. Langridge, Rapid Commun. Mass Spectrom., 1995, 9, 1234 CAS.
- L. E. Edinboro and H. T. Karnes, J. Chromatogr., A, 2005, 1083, 127 CrossRef CAS.
- H. W. Dirr, Biochem Int., 1987, 14(4), 727 CAS.
- L. Tang, M. Tang, L. Xu, H. Luo, T. Huang, J. Yu, L. Zhang, W. Gao, S. B. Cox and J. S. Wang, Carcinogenesis, 2008, 29(2), 411 CAS.
- G. S. Shephard, P. G. Thiel and E. W. Sydenham, J. Chromatogr., A, 1995, 692, 39 CrossRef CAS.
- D. B. Nelson, R. Kimbrough, P. S. Landrigan, A. W. Hayes, G. C. Yang and J. Benanides, Pediatrics, 1980, 66, 865 Search PubMed.
- D. Beker and B. Radic, J. Chromatogr., 1991, 570, 441 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |