A novel C6-phenyl liquid chromatographic technique for rapid and simultaneous measurements of urinary cotinine and nicotine
Bee Lan
Lee
a,
Yanhong
Gao
b,
Ai Li
New
a,
Xu
Wang
a,
Woon-Puay
Koh
a and
Choon Nam
Ong
*ac
aDepartment of Epidemiology and Public Health, Yong Loo Lin School of Medicine, National University of Singapore, 16 Medical Drive, Singapore, 117597. Fax: +65-6779-1489; Tel: +65-6516-4982
bCenter for Disease Control and Prevention of Guangdong Province, 176 Xin Gang Road West, Guangzhou Province, P.R. China 510300
cNUS Environmental Research Institute, National University of Singapore, Singapore
First published on 18th May 2010
Abstract
A simple and rapid liquid chromatographic assay using diode array detection for simultaneous measurements of urinary concentrations of cotinine and nicotine was described. Each urine sample in a volume of 100 μL was extracted with 500 μL of dichloroethane at 60 °C for 5 min. 20 μL of the dried and reconstituted extract was eluted at 1 mL min−1 with a mobile phase at pH 7.6. The urinary cotinine, nicotine and internal standard (indole-3-acetamide) were separated from matrix interferences within 4.2 min on a C6-phenyl column maintained at 50 °C. The calibration curve was linear from 10 to 5000 μg L−1 for cotinine, and from 20 to 7500 μg L−1 for nicotine. The recovery rates of spiked concentrations were close to or about 100% and reproducibility generally greater than 90%. It was observed that the extraction yield of nicotine was very much affected by the extraction temperature. Among various extraction solvents evaluated, dichloroethane offered the highest yield, especially for nicotine. Thus, the urinary nicotine levels determined were generally higher than those reported earlier. This method was used on urine samples from 245 subjects, which included 188 non-smokers, 17 light smokers and 40 heavy smokers. The mean values of cotinine and nicotine were 50 and 96 μg g−1 creatinine among non-smokers, 771 and 679 μg g−1 creatinine among light smokers, and 1422 and 1104 μg g−1 creatinine among heavy smokers.
1. Introduction
Tobacco alkaloids are the active principal components in tobacco products and about 98% of these alkaloid compounds is nicotine.1 Epidemiological studies have shown that tobacco smoking is a major risk factor for cancer, cardiovascular diseases and respiratory illnesses.2–4 In humans, nicotine is mainly metabolized to cotinine and subsequently to trans-3′-hydroxycotinine and other metabolites.5 The measurements of trace levels of nicotine and its metabolites in urine, meconium, hair, saliva or serum have been reported to be useful biomarkers for nicotine metabolism or pharmacokinetics studies.6–8 However, compared to other human biological samples, collection of urine is non-invasive and comparatively easier and more convenient than other methods. The chemical analysis of urine is also easy as the concentrations of nicotine and cotinine are at least 5 folds higher in urine than in plasma or saliva. Hence, urinary nicotine and cotinine concentrations have commonly been used as biomarkers for smoking exposure.9,10 Several analytical techniques for the determination of urinary cotinine and nicotine have been reported in the literature. Immunoassays such as RIA11 and ELISA,12 although cost effective, are considered less specific compared to assays using GC-MS13,14 and HPLC with UV detection,15–17 either alone or coupled with tandem mass spectrometry.18–21 The detection sensitivity of LC-MS/MS has been reported to be 10–40 folds better than UV-mode, but the smaller range of calibration linearity limits its application in urinary analysis for heavy smokers with values possibly >3000 μg L−1. Moreover, the limit of quantification (LOQ) around 20 μg L−1 of nicotine or cotinine obtained from a UV mode may be sufficient for the urinary measurements.22,23 Nevertheless, the extraction variability and problematic resolution of ionizable nicotine on conventional reversed phase column limit the use of such assays to cotinine measurement.24,25In order to overcome the technical difficulties faced by liquid–liquid extraction and RP-C18 chromatographic separation, in this study a novel extraction condition using dichloroethane with temperature control and simple chromatographic assay using a C6-phenyl column with isocratic elution and diode array detection were developed. This method was subsequently used on 245 urine samples collected from a population-based cohort of elderly Chinese in Singapore. Our data showed that urinary nicotine levels were well correlated with cotinine levels, and results obtained from non-smokers were significantly lower than light and heavy smokers. Hence, this method has been proven to be efficient and reliable for routine measurement of urinary levels of nicotine and cotinine as biomarkers of smoking.
2. Experimental
2.1. Reagents and chemicals
(−)-Nicotine (98%), (−)-cotinine (98%) and Indole-3-acetamide (98%) used as internal standard (IS) were obtained from Sigma (St. Louis, MO, USA). Analytical grade of 1,2-dichloroethane was obtained from BDH (Poole, England). Methanol and acetonitrile of HPLC grade were obtained from Tedia (Fairfield, OH, USA). Di-potassium hydrogen phosphate, potassium dihydrogen phosphate, sodium hydroxide and hydrochloric acid, all of pro-analysis grade, were obtained from Merck (Darmstadt, Germany). A buffer solution (pH 11.7) was prepared by mixing 2 mL of 1 M dipotassium hydrogen phosphate and 1 mL of 1 M sodium hydroxide with 7 mL of distilled and deionized water. This buffer was used to condition the pH of urine samples prior to liquid–liquid extraction. (R,S)-Norcotinine, (R,S)-nornicotine, (3′R,5′S)-3′-hydroxycotinine, (1′R,2′S)-nicotine 1,1′-N-oxide, (R,S)-anatabine and (S)-anabasine obtained from Toronto Research Chemicals (North York, ON, Canada) were used to verify the specificity and selectivity of chromatographic determination.2.2. Sample collection and storage
We measured the urinary nicotine and cotinine levels in 245 urine specimens from The Singapore Chinese Health Study, a population-based prospective cohort of middle-aged and elderly Chinese in Singapore.26 Questionnaire data and spot urine used in this study were collected from all consenting participants between 1999 and 2005. For cigarette smoking, the study population was divided into non- and current smokers based on their choice of responses to the following questions, “Have you smoked at least 100 cigarettes in your lifetime” and “Have you smoked at least one cigarette in the past 30 days”. Subjects who answered “yes” to both questions were classified as “current-smokers”, while the rest were classified as “non-smokers”. Current smokers were then asked about number of cigarettes smoked per day and categorized as light (1–12 cigarettes per day) or heavy smokers (more than 12 cigarettes per day). Spot urine specimens were collected from the participants and preserved with ascorbic acid prior to the storage at −80 °C. Urine samples from 3 volunteers (one non-smoker, one light and one heavy smoker) working in the university were also collected and used as internal controls. The urine sample of the non-smoking volunteer was used as blank and spiked with tested compounds for recovery and reproducibility studies. The two other urine samples from the light and heavy smokers were used as quality control samples (QC1 and QC2), for the entire study. One mL from each of the above three urine samples was mixed with 10 μL of 6 M hydrochloric acid in micro-centrifuge tubes and kept at −20 °C until analysis.2.3. HPLC apparatus and conditions
The apparatus used was a Shimadzu Prominence LC-20AT binary pumping system coupled with a CTO-10A column oven, SPD-M20A diode array detector and a CBM-20A communication bus module that was controlled by LC Solution Chromatography Data System (Kyoto, Japan). The analytical column used was a Phenomenex Gemini C6-phenyl column (5 μm, 50 × 4.6 mm i.d.) protected by a C6-phenyl security guard cartridges (4 × 3 mm i.d., Torrance, CA, USA). The column temperature was maintained at 50 °C. A mobile phase with pH 7.6 ± 0.05 was a mixture of 135 mL of 100 mM dipotassium hydrogen phosphate, 65 mL of 100 mM potassium dihydrogen phosphate, 80 mL of methanol and 90 mL of acetonitrile and 630 mL of distilled and deionized water. The flow rate was 1 ml min−1. The analysis was initiated immediately after an auto-injection of sample (Gilson Model 234 auto-injector, Villiers-le-Bel, France). Using the diode array detection, the target compounds were determined at 260 nm and the peak resolutions were re-confirmed with peak purity test.2.4. Calibrators, quality controls and spiked urine samples
The stock solutions of nicotine, cotinine and IS, each 1 mg mL−1, were prepared individually in methanol–water, 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (v/v). A working IS solution (2 mg L−1) used for calibration and the standard mixtures with various concentrations ranging from 40 to 15000 μg L−1 of nicotine and 20 to 10000 μg L−1 of cotinine, were all prepared in mobile phase solution. An aliquot of 50 μL from each standard mixture was mixed with 50 μL of IS solution, and 20 μL of each of these calibrators was used for chromatographic analysis. To assess the precision and accuracy of the method, the urine sample from the non-smoking volunteer was spiked with various concentrations as listed in Table 1. Five assays each from the blank and 3 spiked samples were performed within a day and repeated in five consecutive days. For quality control purpose, samples measured in the daily analysis always included duplicate assays of the two QC samples.
:2 (v/v). A working IS solution (2 mg L−1) used for calibration and the standard mixtures with various concentrations ranging from 40 to 15000 μg L−1 of nicotine and 20 to 10000 μg L−1 of cotinine, were all prepared in mobile phase solution. An aliquot of 50 μL from each standard mixture was mixed with 50 μL of IS solution, and 20 μL of each of these calibrators was used for chromatographic analysis. To assess the precision and accuracy of the method, the urine sample from the non-smoking volunteer was spiked with various concentrations as listed in Table 1. Five assays each from the blank and 3 spiked samples were performed within a day and repeated in five consecutive days. For quality control purpose, samples measured in the daily analysis always included duplicate assays of the two QC samples.
2.5. Sample preparation
An IS-buffer solution that containing 2000 μg L−1 of IS was freshly prepared by diluting the IS stock solution with 500-fold of the pH conditioning buffer (pH 11.7). After urine samples were properly thawed, mixed and centrifuged at 12700 rpm for 2 min, an aliquot of 100 μL of urine from each sample was added with 50 μL IS-buffer solution and 500 μL of dichloroethane. A batch of 8 samples was placed in a Vortemp™ vortexing incubator (UniEquip, Germany) with temperature set at 60 °C and shaking speed at 1350 rpm. After extraction for 5 min, the sample was centrifuged at 12700 rpm for 30 s and 450 μL of the organic phase was immediately transferred for drying. With the use of Supelco Visidry™ and Visprep™ devices, these samples were dried together under nitrogen stream for 15–20 min. The dried residue was reconstituted in 90 μL of mobile phase and 20 μL was used for chromatographic analysis. The results were presented as observed data (μg L−1) or data after correction for urinary creatinine (μg g−1 creatinine).
2.6. Statistical analysis
Urinary cotinine and nicotine levels were compared among non-smokers, light and heavy smokers using the one-way ANOVA tests followed by post-hoc paired wise comparisons using Bonferroni tests.3. Results and discussion
3.1. Chromatographic separation and efficiency
In the present study, using C18 chromatography, nicotine was mainly present in its ionized form under acidic condition and the stationary phase was transient. Thus, the measurement of nicotine could hardly be achieved without using ion pairing reagent, and the resolution of this compound had to be improved by adding triethylamine into the mobile phase.15,17 Hence, the conditions reported earlier using C18 chromatography was only useful for sole quantification of urinary cotinine.24,25 In our present study, it was observed that the retention for nicotine was best for the mobile phase of neutral pH. With the use of a C6-phenyl stationary phase that is stable under a wide range of mobile phase pH from 1 to 12, nicotine could be eluted using medium alkaline carrier, and resolved from matrix interference. It was interesting to observe that increase in acetonitrile concentration in the mobile phase led to faster elution of the tested compound. However, the increase in column temperature and methanol content did not have the same effect on nicotine. Among the 3 C6-phenyl columns of different dimensions (100 × 4.6 mm i.d., 50 × 4.6 mm i.d. and 50 × 3 mm i.d.) tested in our laboratory, column of 50 × 4.6 mm i.d. was observed to be the best as it offered both excellent resolution and short chromatographic run time.The present HPLC conditions, with column temperature maintained at 50 °C, mobile phase pH at 7.6, and both acetonitrile and methanol as organic modifiers, offered the best separation of target compounds on C6-phenyl column. As shown in Fig. 1A, the analytes of interest were well separated from the other minor metabolites of nicotine (norcotinine, nornicotine, 3-hydroxycotinine, nicotine oxide) and possible components of tobacco products (anatabine, anabasine) with similar spectrum and peak apex at around 260 nm. The retention times for cotinine (2500 μg L−1), IS (1000 μg L−1) and nicotine (5000 μg L−1) were 1.91 min, 3.19 min and 4.06 min, respectively. The retention times were highly reproducible, with between-day variation <0.3%. The LOQ of the assays (S/N = 5) is shown in the Fig. 1B. The lowest detectable level of cotinine in urine was 10 μg L−1 and 20 μg L−1 for nicotine. Chromatograms of urine samples collected from the non-smoking volunteer and spiked with 1000 μg L−1 of cotinine and 2000 μg L−1 of nicotine are shown in Fig. 1C and 1D, respectively. The chromatogram for the quality control sample QC2 obtained from the heavy smoker with measured cotinine 1743 μg L−1 and nicotine 6033 μg L−1 is shown in Fig. 1E. As demonstrated in all chromatograms, the separations of target compounds were free from matrix interferences.
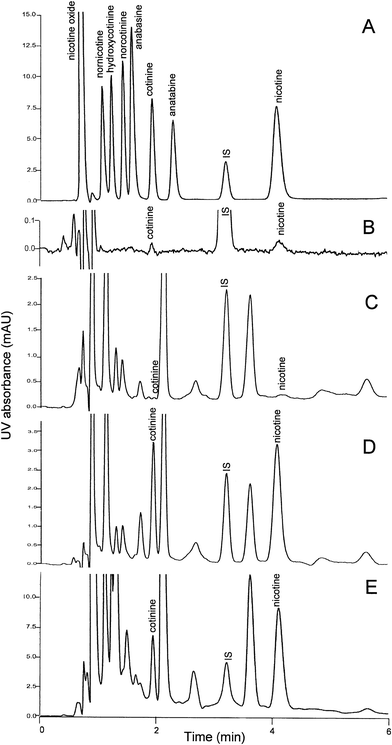 | ||
| Fig. 1 Chromatograms extracted from DAD spectra at 260 nm: (A) The pure standard mixtures of cotinine (2500 μg L−1), nicotine (5000 μg L−1), IS (1000 μg L−1) and 2500 μg L−1 each of norcotinine, nornicotine, 3-hydroxycotinine, nicotine oxide, anatabine and anabasine; (B) the lowest detectable concentrations of cotinine, 10 μg L−1 and nicotine, 20 μg L−1; (C) a blank with 18 μg L−1 of cotinine and 42 μg L−1 of nicotine determined; (D) its spiked urine samples with added 1000 μg L−1 of cotinine and 2000 μg L−1 of nicotine and (E) a QC sample collected from heavy smoker with concentration of cotinine measured as 1743 μg L−1 and nicotine as 6033 μg L−1. | ||
3.2. Extraction and detection efficiency
Several GC-MS13,14 and LC-MS/MS18,19 methods for cotinine and nicotine determinations have been described in the literature. Nevertheless, these assays required over 1 mL of urine sample for liquid–liquid or solid-phase extractions prior to chromatographic determination. Although the latest automated in-tube solid-phase micro-extraction coupled with LC-MS assay21 could be more convenient than the off-line extraction process, the setback of this assay was the sophisticated procedures of 25 draw/eject cycles with 40 μL of 10-fold diluted urine sample at a critical flow rate. Furthermore, the sensitivities achieved by MS far exceeded the requirement for measurements of cotinine and nicotine levels in human urine. A UV-based method that enable the quantification of around 20 μg mL−1 of cotinine is sufficient to distinguish non-smokers from passive, minimally and highly active smokers with their cut-off points recommended as 50, 170, 550 and 2100 μg L−1, respectively.23 Moreover, detection using UV is comparatively simple and cost-effective for large scale population study. A common disadvantage of the earlier UV-based methods was the tedious and time consuming procedures of drying and reconstitution of large amount of urine used for the assay. This was because small sample volumes and low extraction yield did not provide adequate detection sensitivity. Hence, most assays generally required 5 to 20 mL of urine sample.16,24,25 In our preliminary study, it was noted that the extraction yield of nicotine was much lower than cotinine when the sample was treated with chloroform, and this finding was similar to several earlier reports.14,16,27 Further, it was also noted that the nicotine level presented in some urine samples which were extracted with different solvents could be as high as or even higher than cotinine level in some smokers. Thus, we decided to examine the efficiency of liquid–liquid extraction method using 6 different types of organic solvent. The results are shown in Table 2. Among the tested solvents, dichloromethane offered the highest recovery, although the reproducibility was more promising with the use of the dichloroethane. This could be due to the fact that pipetting of dichloroethane with an air displacement pipettor, which was much easier than with dichloromethane, since the latter is of higher specific gravity and volatility. On the other hand, it is also interesting to note that the physico-chemical property of nicotine is unlike cotinine; nicotine is miscible at aqueous phase when temperature was lower than 60 °C. The quality control sample QC2 obtained from the heavy smoker was used to study further the effect of temperature changes during the extraction using the three best extraction solvents. It was observed that dichloroethane (with a higher boiling point at 83 °C) was found to be the most effective extraction solvent, with the highest extraction yield at 55–60 °C. The extraction yield ratio (Y-axis) in Fig. 2 was calculated based on the peak height of individual analytes obtained from various conditions against the peak height of the same compound extracted at 60 °C with dichloroethane. The results suggest that the extraction yields of target compounds especially for nicotine were very much affected by the extraction solvent and extraction temperature. These studies suggest that reliable measurements could be achieved on 100 μL of urine sample with a single step extraction using dichloroethane. This liquid–liquid extraction procedure allowed a batch of 8 samples to be treated within 30 min.| Solvents | Mean extraction yield ± SD (%) | ||
|---|---|---|---|
| Nicotine | Cotinine | IS | |
| Dichloromethane | 89 ± 9 | 94 ± 7 | 75 ± 7 |
| Dichloroethane | 85 ± 1 | 85 ± 3 | 74 ± 3 |
| Chloroform | 71 ± 9 | 75 ± 8 | 57 ± 8 |
| Trichloroethylene | 66 ± 10 | 34 ± 3 | 12 ± 1 |
| Diethyl ether | 47 ± 3 | 19 ± 11 | 51 ± 10 |
| Hexane | 35 ± 10 | 1 ± 0.1 | 1 ± 0.4 |
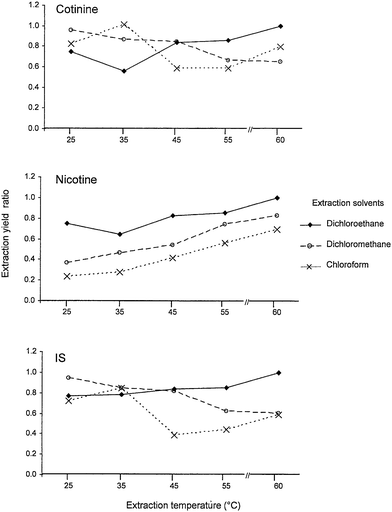 | ||
| Fig. 2 The temperature effect on the dichloroethane, dichloromethane and chloroform extraction yields of cotinine, nicotine and IS (Extraction yield ratio = 1 was defined for the optimum condition of dichloroethane extraction at 60 °C). | ||
3.3. Calibration, recovery and reproducibility
The calibration curves were constructed routinely using the known concentrations (y) ranged from 10 to 5000 μg L−1 for cotinine and 20 to 7500 μg L−1 for nicotine plotted against their peak height ratios (x) to the internal standard method. Linear regression analysis of the calibration curves resulted in the following equations: y = 891x − 1, r = 0.99 for cotinine and y = 1895x − 2, r = 0.99 for nicotine. The between-day variations (n = 5) of the slopes and linearity were generally <2% and <0.01%, respectively. The actual recovery rates based on the direct peak height comparison for added cotinine and nicotine concentrations were around 85%, and for IS was 74%. With the use of internal standard method, the adjusted recovery rate ranged from 106–113%, the coefficients of variations (n = 5) of within-day assays and between-day assays were generally <8% for cotinine and <11% for nicotine (Table 1).3.4. Urinary concentrations of nicotine and cotinine
Cigarette smoking has been known for years to impair human health. Nicotine, presents in mainstream and sidestream smoke, is believed to be one of the most toxic components of tobacco. Cotinine, which has a 20-hour half-life is thought to be an inactive metabolite of nicotine as its elimination profile is more stable than that of nicotine. As a result, cotinine is considered a good biological marker for determining smoking exposure.10,28 Nevertheless, it is believed that urinary nicotine should also be considered as an additional biomarker to complement urinary cotinine. A total of 245 urine specimens were analyzed using the proposed method by chromatographers who were blinded to the subjects' smoking status and related information. The overall results showed that urinary nicotine levels correlated closely with cotinine levels (coefficient of correlation r = 0.77, p < 0.001). Our analysis showed that there were significant differences in urinary nicotine and cotinine levels between smokers and non-smokers, and also a positive association between the levels of nicotine and cotinine, and the dosage of smoking. The distribution of urinary cotinine and nicotine levels in these 245 samples is shown in Fig. 3. The mean urinary cotinine levels among 188 non-, 17 light and 40 heavy smokers were 50, 771 and 1422 μg g−1 creatinine, respectively. The corresponding values for urinary nicotine levels were 96, 679 and 1104 μg g−1 creatinine. The difference in levels among the three groups was statistically significant for both cotinine (p < 0.001) and nicotine (p = 0.028).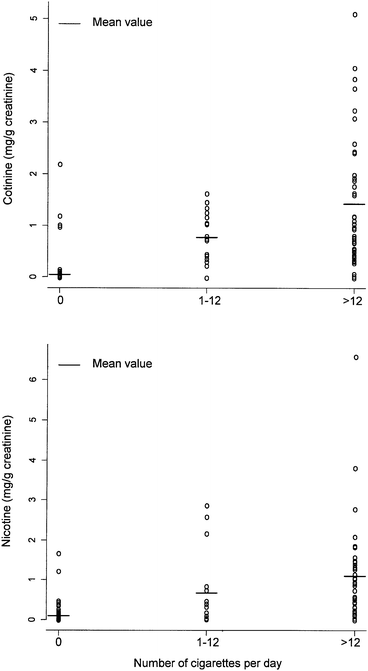 | ||
| Fig. 3 Distribution of urinary cotinine and nicotine levels in 245 study samples (188 non-, 17 light and 40 heavy smokers). | ||
4. Conclusion
This paper shows that the LC-UV method could be best performed using a C6-phenyl stationery phase instead of RP-C18 or C8. The extraction yield, especially for urinary nicotine, was found to be affected by extraction solvent and temperature. The proposed method is shown to be sensitive, simple, rapid and cost-effective. Eight urine samples can be prepared in less than half an hour and more than 500 samples can be measured within 5 consecutive days. This method is useful for large scale population study for risk assessment of tobacco smoke exposure.Acknowledgements
This study was carried out with the support of the Singapore Biomedical Research Council (BMRC, grant No. R-186-000-068-305) and the Life Sciences Institute Toxicology Program. The Singapore Chinese Health Study was funded by National Institutes of Health (NCI R01 CA55069, R35 CA53890 and R01 CA80205).References
- M. V. Djordjevic and K. A. Doran, Handb. Exp. Pharmacol., 2009, 192, 61–82 Search PubMed.
- J. M. Yuan, W. P. Koh, S. E. Murphy, Y. H. Fan, R. W. Wang, S. G. Carmella, S. M. Han, K. Wickham, Y. T. Gao, M. C. Yu and S. S. Hecht, Cancer Res., 2009, 69, 2990–2995 CrossRef CAS.
- J. Finkelstein, E. Cha and S. M. Scharf, Int. J. Chron. Obstruct. Pulmon. Dis., 2009, 4, 337–349 Search PubMed.
- S. Pavanello, P. Simioli, M. Carrieri, P. Gregorio and E. Clonfero, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2002, 521, 1–9 CrossRef CAS.
- J. Hukkanen, P. Jacob and N. L. Benowitz, Pharmacol. Rev., 2005, 57, 79–115 CrossRef CAS.
- M. Simoni, S. Baldacci, R. Puntoni, F. Pistelli, S. Farchi, E. Lo Presti, R. Pistelli, G. Corbo, N. Agabiti, S. Basso, G. Matteelli, F. Di Pede, L. Carrozzi, F. Forastiere and G. Viegi, Clin. Chem. Lab. Med., 2006, 44, 632–638 CrossRef CAS.
- C. S. Parzynski, M. Jaszyna-Gasior, F. H. Franken and E. T. Moolchan, Pharmacol., Biochem. Behav., 2008, 89, 145–149 CrossRef CAS.
- N. L. Benowitz, J. Hukkanen and P. Jacob, Handb. Exp. Pharmacol., 2009, 192, 29–60 Search PubMed.
- A. Florescu, R. Ferrence, T. Einarson, P. Selby, O. Soldin and G. Koren, Ther. Drug Monit., 2009, 31, 14–30 CrossRef CAS.
- N. L. Benowitz, K. M. Dains, D. Dempsey, B. Herrera, L. Yu and P. Jacob, Nicotine & Tobacco Research, 2009, 11, 954–960 Search PubMed.
- K. L. Chambers, G. A. Ellard, A. T. Hewson and R. F. Smith, Br. J. Biomed Sci., 2001, 58, 61–65 Search PubMed.
- T. Wielkoszynski, K. Tyrpien and M. Szumska, J. Pharm. Biomed. Anal., 2009, 49, 1256–1260 CrossRef CAS.
- A. J. Ji, G. M. Lawson, R. Anderson, L. C. Dale, I. T. Croghan and R. D. Hurt, Clin. Chem., 1999, 45, 85–91 CAS.
- C. N. Man, L. H. Gam, S. Ismail, R. Lajis and R. Awang, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2006, 844, 322–327 CrossRef CAS.
- M. Hariharan and T. Vannoord, Clin. Chem., 1991, 37, 1276–1280 CAS.
- C. Oddoze, A. M. Pauli and J. Pastor, J. Chromatogr., B: Biomed. Sci. Appl., 1998, 708, 95–101 CrossRef CAS.
- A. M. Maissadeh, A. A. Gharaibeh and K. W. Omari, J. Chromatogr., Sci., 2009, 47, 170–177.
- X. Xu, M. M. Iba and C. P. Weisel, Clin. Chem., 2004, 50, 2323–2330 CrossRef CAS.
- A. N. Hoofnagle, T. J. Laha, P. M. Rainey and S. M. Sadrzadeh, Am. J. Clin. Pathol., 2006, 126, 880–887 CrossRef CAS.
- C. A. Chadwick and B. Keevil, Ann. Clin. Biochem., 2007, 44, 455–462 CrossRef CAS.
- H. Kataoka, R. Inoue, K. Yagi and K. Saito, J. Pharm. Biomed. Anal., 2009, 49, 108–114 CrossRef CAS.
- S. L. Bramer and B. A. Kallungal, Biomarkers, 2003, 8, 187–203 CrossRef CAS.
- W. Zielinska-Danch, W. Wardas, A. Sobczak and I. Szoltysek-Boldys, Biomarkers, 2007, 12, 484–496 CrossRef CAS.
- F. Ceppa, Y. El Jahiri, H. Mayaudon, O. Dupuy and P. Burnat, J. Chromatogr., B: Biomed. Sci. Appl., 2000, 746, 115–122 CrossRef CAS.
- L. Rabbaa-Khabbaz, R. A. Daoud and D. Karam-Sarkis, J. Chromatogr., Sci., 2006, 44, 535–538 CAS.
- W. P. Koh, J. M. Yuan, C. L. Sun, H. P. Lee and M. C. Yu, J. Nutr., 2005, 135, 2473–2477 CAS.
- C. N. Man, A. I. Fathelrahman, G. L. Harn, R. Lajis, A. S. M. Samin, M. Omar, R. Awang and N. A. Bayanuddin, Environ. Toxicol. Pharmacol., 2009, 28, 92–96 CrossRef CAS.
- S. W. Park and J. Y. Kim, Journal of Preventive Medicine and Public Health, 2009, 42, 223–230 Search PubMed.
| This journal is © The Royal Society of Chemistry 2010 |