
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInexpensive and bench stable diarylmethylium tetrafluoroborates as organocatalysts in the light mediated hydrosulfonylation of unactivated alkenes†
Polyssena
Renzi
 ,
Emanuele
Azzi
,
Sylvain
Ascensio
,
Stefano
Parisotto
,
Fabrizio
Sordello
,
Francesco
Pellegrino
,
Giovanni
Ghigo
* and
Annamaria
Deagostino
*
,
Emanuele
Azzi
,
Sylvain
Ascensio
,
Stefano
Parisotto
,
Fabrizio
Sordello
,
Francesco
Pellegrino
,
Giovanni
Ghigo
* and
Annamaria
Deagostino
*
Department of Chemistry, University of Torino, Via Pietro Giuria, 7, 10125 Torino, Italy. E-mail: deagostino.annamaria@unito.it; giovanni.ghigo@unito.it
First published on 10th February 2023
Abstract
In this paper, we present the synthetic potential of diarylmethylium tetrafluoroborates as catalysts for the visible light promoted hydrosulfonylation of unactivated alkenes. For the first time, these salts, which are bench stable and easily preparable on a multi-gram scale, were employed as organocatalysts. Interestingly, a catalyst loading of only 1 mol% allowed sulfone products to be efficiently obtained from good-to-excellent yields with high functional-group tolerance and scalability up to 15 mmol of alkene. The mechanistic study, both experimental and computational, presented here, revealed an alternative mechanism for the formation of the key sulfonyl radical. Indeed, the photoactive species was proved not to be the diarylcarbenium salt itself, but two intermediates, a stable S–C adduct and an ion couple, that were formed after its interaction with sodium benzenesulfinate. Upon absorbing light, the ion couple could reach an excited state with a charge-transfer character which gave the fundamental sulfonyl radical. A PCET (proton-coupled electron transfer) closes the catalytic cycle reforming the diarylcarbenium salt.
Introduction
Sulfones are privileged building blocks for medicinal chemistry, drug discovery and natural-product synthesis,1,2 therefore, the search for operationally simple and easily scalable methodologies for their preparation is a current hot topic.3,4 To overcome the drawbacks associated with the traditional oxidation of sulfides or sulfoxides,5–8 considerable effort has been devoted to the development of SO2 surrogates,9–11 and to the employment of alternative solvents (water, deep eutectic solvents, and solvent free).12,13 Moreover, emerging techniques such as photochemistry14,15 and electrochemistry are increasingly gaining ground.12 In most cases, the key step to the success of these reactions is the generation of the sulfonyl radical from suitable precursors (e.g. sodium sulfinates, sulfonyl chlorides, sulfinic acids, sulfonyl hydrazides, sulfonyl imines, and DABCO(SO2)2).16–20 Vicinal difunctionalisation is generally observed under photocatalysis21–24 when feedstock chemicals, such as alkenes, are employed as coupling partners, while methods enabling their direct hydrosulfonylation are very limited. The first example was reported by Yu in 2019 (Scheme 1a).25 The anti-Markovnikov hydrosulfonylation of alkenes bearing alkyl groups was accomplished under iridium-photocatalysis in the presence of sodium sulfinates, used as sulfonyl radical precursors. This methodology has also been applied to electron-poor alkenes showing reduced reactivity and/or the formation of product mixtures. In 2020, Liu and Duan applied the protocol to styrenes by substituting acetic acid and water with methanol for use as a proton source (Scheme 1b).26 Pre-activated sulfone tetrazoles, and activated sulfonyl chlorides were also proposed as sulfonyl radical precursors by Ley27 and Gouverneur.28 In the first case, acrylates and electron-poor alkenes can be readily converted to dialkyl sulfones under iridium photoredox catalysis. The success of this protocol is somewhat tempered by the need for a stoichiometric amount of DMAP as a sacrificial reagent, and Cu(OAc)2 as an external oxidant (Scheme 1c). In the second case, sulfonyl chlorides are employed for the hydrosulfonylation of electron-deficient alkenes under visible light activation for the first time. However, the use of (TMS)3-SiH as a H donor is necessary. In this way, they have developed a mild protocol that is suitable for late-stage functionalisation (Scheme 1d).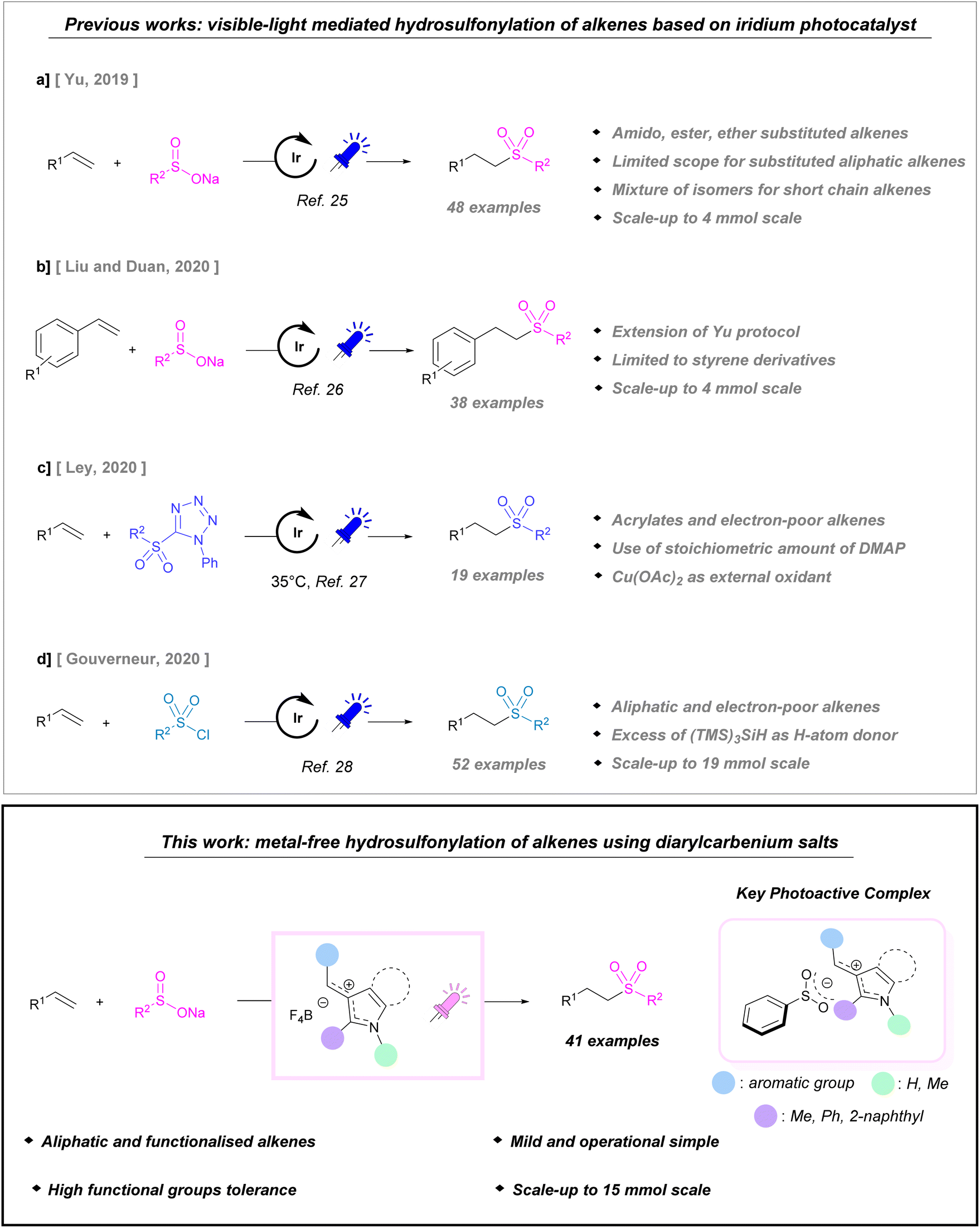 | ||
| Scheme 1 Top: previously reported visible-light hydrosulfonylation of alkenes; bottom: this work. | ||
More recently, Xue and Luo have reported the use of sulfonyl imines as versatile sulfonyl precursors under iridium/copper dual catalysis.20 As shown in Scheme 1, the hydrosulfonylation of alkenes mediated by visible light is dominated by iridium catalysis. Although polypyridyl complexes of iridium are robust photocatalysts, they can be acid sensitive29 and the low abundancy of this metal30 makes these compounds unsustainable from economic and environmental points of view. Moreover, their high costs potentially limit their application on industrial scale. While iridium and ruthenium complexes still stand at the forefront of photocatalysis, in recent years, organic dyes (e.g. acridinium salts, anthraquinones, Eosin Y, cyanoarenes, etc.) have emerged as a sustainable option due to their stability, photocatalytic performances and wide range of useful redox potentials.31–36
As part of our ongoing research on the photocatalyzed functionalisation of unsaturated compounds,37–39 we have developed an operationally simple anti-Markovnikov hydrosulfonylation of a broad range of unactivated alkenes 1 with sodium sulfinates 2a–e in the presence of diarylmethylium tetrafluoroborates under purple light (Scheme 1, bottom). This class of compounds is known in the literature as a model substrate in organocatalyzed alkylation reactions40–43 or together with benzhydrylium ions have been employed by Mayr as reference electrophiles for the construction of a general basicity scale.44,45 For the first time, we have employed non-symmetric diarylcarbenium salts I–XII as organocatalysts in a light mediated process. These salts strongly absorb in the visible spectrum, with absorption maxima ranging from red to yellow according to their substitution patterns. They can be easily synthesised on the gram scale via the direct condensation of aryl or heteroaryl aldehydes and N-heteroarenes. Interestingly, they are bench-stable salts with long shelf life and high stability, embodying the characteristics of the ideal catalyst. Furthermore, their structure can be easily modulated to finely tune their redox potentials. An additional aspect that should not be underestimated is the catalyst cost as diarylcarbenium salts are more economically viable than iridium catalysts.46
Results and discussion
We initiated our study by evaluating the spectroscopic and electrochemical properties of several diarylcarbenium tetrafluoborates I–XII, which were available in our laboratory (for full characterization see Table S7 in the ESI†). Cyclic voltammetry (CV) measurements showed an interesting match between the redox potentials of salts I–XII and sodium benzenesulfinate 2a, which suggested their potential application as a catalyst in the hydrosulfonylation of unsaturated compounds. Therefore, hexadec-1-ene 1a and sodium benzenesulfinate 2a were chosen as the model substrates to prove our hypothesis, starting from reaction conditions reported by Yu.25 The reaction was not feasible at room temperature, at 40 °C or under illumination at 525 nm (Table 1 entries 2–3) in the presence of tetrafluoroborate salt I (10 mol%), water (10 eq.) and acetic acid (4.5 eq.) in CH2Cl2, whereas product 3a was isolated in a 22% yield under illumination with a 40W Kessil blue LED lamp (Table 1 entry 4). Unsatisfactory results were obtained under all the conditions tested at 456 nm (for the complete set of experiments see Table S1†). This observation and the lack of reactivity under green light can be rationalized by the colour bleaching of diarylcarbenium I when sulfinate 2a was added to the reaction medium, which hints at there being a different active species than I in this light-mediated process.|
|
||
|---|---|---|
| a Reaction conditions: hexadec-1-ene 1a (0.5 mmol), sodium benzenesulfinate 2a (0.8 mmol, 1.6 eq.), I–XII (1 mol%), in CH2Cl2 (2.5 mL; 0.2 M), CH3COOH (2.25 mmol, 4.5 eq.), H2O (5 mmol, 10 eq.), 40W Kessil purple LED lamp (390 nm), 23 h, room temperature. b Determined on the isolated product. c Carried out with 10% mol of I. | ||
| a. Optimisation of the reaction conditions | ||
| Entry | Deviation | 3a Yield [%]b |
| 1 | None | 80 |
| 2 | No irradiation at rt or 40 °C | 0 |
| 3 | Green LED lamp (525 nm) | 0 |
| 4 | Blue LED lamp (456 nm) | 22c |
| 5 | I (10 mol%) | 71 |
| 6 | I (5 mol%) | 87 |
| 7 | I (3 mol%) | 83 |
| 8 | No I | 0 |
| 9 | CHCl3 as solvent | 50 |
| 10 | THF as solvent | 50 |
| 11 | CH3CN as solvent | 30 |
| 12 | 0.1 M in CH2Cl2 | 64 |
| 13 | No acid | Traces |
| 14 | HCOOH as acid | 77 |
| 15 | 1 eq. of CH3COOH as acid | 41 |
| 16 | No H2O | 0 |
| 17 | H2O (6 eq.) | 66 |
| 18 | H2O(20 eq.) | 72 |
| 19 | 2a (1.1 eq.) | 59 |
| 20 | 2a (3 eq.) | 70 |
| b. Screening of selected diarylmethylium tetrafluoroborates salts | ||
|
||

Therefore, we tested the hydrosulfonylation under purple LED lamp irradiation (390 nm) and we were delighted to observe a 71% yield of 3a in the presence of 10 mol% of I (Table 1 entry 5). As described in Table 1 entry 6, a decrease in the catalyst amount, from 10 to 5 mol%, was beneficial to the reaction outcome with an increase in yield up to 87%. A further reduction in I, loading at 3 mol%, caused a slight decrease to an 83% yield (Table 1 entry 7). Interestingly, the use of only 1 mol% of salt I still resulted in an 80% yield (Table 1 entry 1). We consider this to be the most convenient result because a high yield was obtained with a very low loading of I, which is an organocatalyst. Moreover, this amount corresponds to the catalyst loading generally used in iridium photocatalysis in similar reactions.25,26 We therefore decided to continue our optimization with 1 mol% of I. We also confirmed a lack of reactivity under illumination with a 390 nm LED lamp in the absence of the diarylcarbenium salt I (Table 1 entry 8).
Then, reaction conditions were extensively screened in order to assess the role of the solvent, acid, water and sulfinate amount (see the ESI† for exhaustive Tables). Regarding the solvent, the protic CHCl3 and the aprotic THF were less efficient than CH2Cl2 both affording the product with a 50% yield (Table 1 entries 9–10). The use of CH3CN caused a further drop to a 30% yield (Table 1 entry 11). Other solvents such as DMF or 2-Me-THF proved to be very inefficient (see Table S3†). A dilution of the reaction mixture from 0.2 M to 0.1 M in CH2Cl2 was not beneficial, resulting in a 64% yield of 3a (Table 1 entry 12).
The reaction outcome was highly dependent on the presence of the acid and water with a complete lack of reactivity in the absence of both (Table 1 entries 13 and 16). Organic acids such as formic and acetic acid (Table 1 entries 14 and 1, respectively 77 and 80%) performed better than inorganic acids, the latter causing the degradation of alkene 1a (for the complete screening see Tables S4 and S5†). A screening of the equivalents of acetic acid was also accomplished, showing the need for an excess of this acid. As reported in entry 15 of Table 1, the use of a stoichiometric amount of acetic acid decreased the yield to 41% (versus 80% yield with 4.5 eq. of acetic acid). An excess of water was necessary, in order to assure the best solubility of sodium benzenesulfinate 2a. Best results were obtained with 10 equivalents as shown in Table 1 entry 1. Higher or lower amounts resulted in a slight decrease in the reaction efficiency, a 66% and 72% yield for 3a were respectively observed with 6 and 20 equivalents of water (Table 1 entries 17–18, see also Table S5†). Our attention was then pointed towards the influence of the sodium benzenesulfinate 2a amount with respect to hexadec-1-ene 1a. Neither the use of 1.1 (59%, Table 1 entry 19) or 3 equivalents of 2a (70%, entry 20) resulted to be beneficial in comparison to the starting 1.6 equivalents.
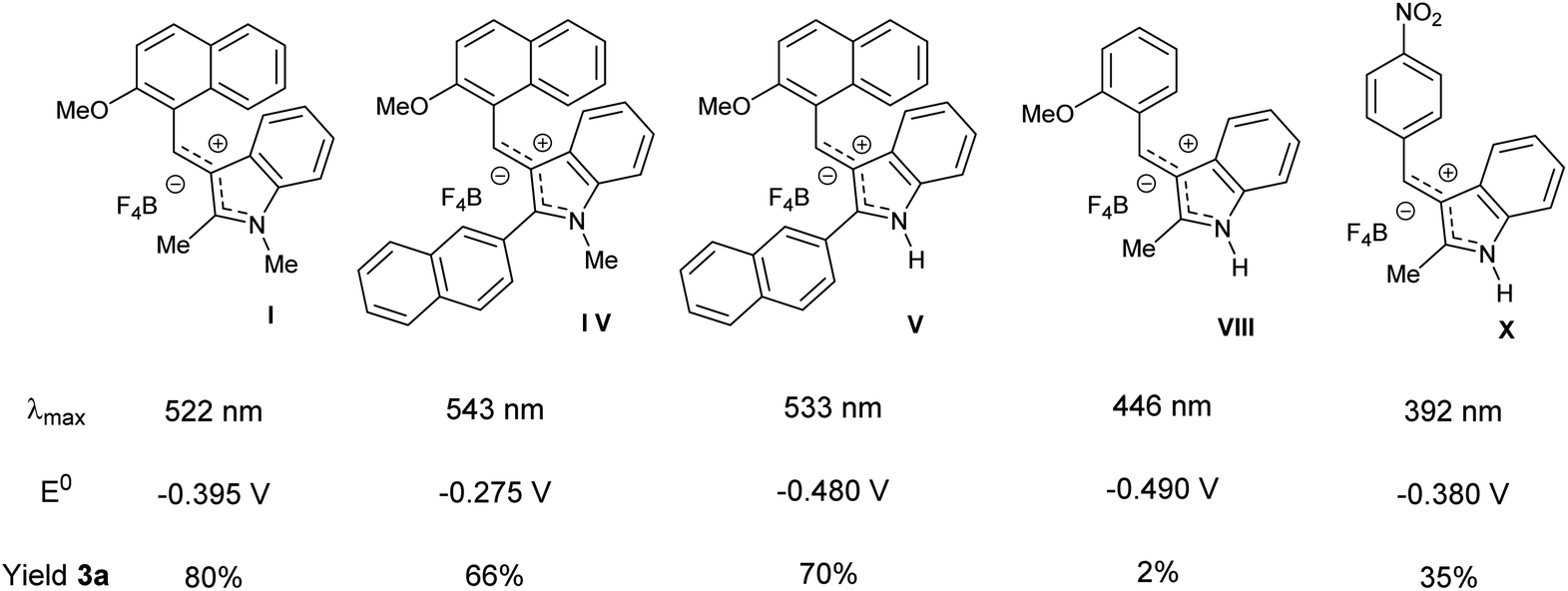
We next sought to evaluate the catalyst structure changing the aryl moiety and the substituents on C2 and nitrogen of the indole portion of the diarylcarbenium salt. A selection of the results is reported in Table 1b, whereas the complete study is described in Table S7 of the ESI.† According to the substitution patterns, the diarylcarbenium salts I–XII showed a different efficiency in promoting the hydrosulfonylation of hexadec-1-ene 1a with 2a. Several tetrafluoroborates were prepared and the effect of the substituents at C2 and N of the indole core was evaluated. In general, from good to high yields were obtained with diarylcarbenium salts I–IV, bearing a methoxy-naphthyl moiety and a N-methyl indole. A moderate decrease in yield from 80 to 66% was observed increasing the size of the substituent at C2 of the indole portion while keeping the 2-methoxynaphthyl moiety (Table 1b, I and IV). A dramatic drop in the efficiency was achieved for those catalysts derived from a substituted benzaldehyde and 2-methylindole (Table 1b, VIII and X). Therefore, the presence of a methylated nitrogen proved to be essential for high yield except for compound V which despite being a N–H salt gave a 70% yield for 3a (Table 1b).
Although most diarylcarbenium tetrafluoborate salts showed discrete to good efficiency, no direct correlation could be found with their electronic properties and redox potentials. As shown in Table 1b, tetrafluoroborate salts with similar redox potentials, e.g. I and X or V and VIII, gave very different yields in the hydrosulfonylation of hexadec-1-ene 1a. Therefore, we became suspicious of the role of diarylmethylium tetrafluoborates as effective catalysts in this transformation. We must recall also the colour bleaching observed upon the addition of 2a. Mechanistic studies were then initiated in order to understand the actual catalytic species involved here.
In summary, the complete screening of catalysts, reagent amounts and ratios (fully described in the ESI†) confirmed the conditions reported in Table 1 entry 1 to be the optimal. Thus, a simple setup composed of 1 mol% of salt I, 4.5 equivalents of acetic acid and 10 equivalents of water in CH2Cl2 under irradiation with a purple LED lamp at room temperature enabled an efficient anti-Markovnikov functionalisation of alkene 1a to afford sulfone 3a.
Mechanistic studies: experimental and computation
Experimental mechanistic studies
In order to understand the function of purple light in the hydrosulfonylation of alkenes, we turned our attention to the reaction mechanism. The crucial role of light was demonstrated by a light/dark experiment which showed reaction progression solely under irradiation (Fig. 1a). Then, to gain further insight into the reaction mechanism and prove the formation of radical intermediates, we performed trapping experiments with 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) and butylhydroxytoluene (BHT) as radical scavengers (Fig. 1b). When 2 equivalents of TEMPO were added to the reaction mixture under standard conditions, the transformation efficiency drastically dropped (16% yield vs. 80% under standard conditions for 3a). In the presence of the radical scavenger BHT, the yield of the sulfonylation product 3a further dropped to 10% suggesting that a radical pathway could be involved in this transformation (Fig. 1b). This idea was additionally confirmed by the detection of the BHT-sulfonyl radical adduct 5 by HRMS. Furthermore, when the reaction was carried out under an O2 atmosphere, no product formation was observed. The reaction proceeded with an anti-Markonikov selectivity in which acetic acid played a crucial role, as shown in the optimisation. Moreover, deuterium experiments showed the introduction of deuterium into the β-position of sulfone 3a. The deuterated product 3a′ was obtained along with its non-deuterated counterpart 3a in 47/53 ratio in the presence of CD3COOD and D2O (Fig. 1c). | ||
| Fig. 1 a) The light/dark experiment. (b) The TEMPO/BHT trapping experiment. (c) The deuterium experiment. The details of the experimental conditions for each study are reported in the ESI.† | ||
Determination of the catalytic species
In order to determine the actual catalytic species, we performed the UV-vis analysis of diarylmethylium salt I alone and in the presence of sodium benzenesulfinate 2a. As shown in Fig. 2a, salt I is a bright coral red solid showing two absorption bands centered at 522 (visible) and 382 (UV-A) nm (black line in 2a) in CH2Cl2. When sodium benzenesulfinate 2a is added to I solution, a fast and progressive colour fading to colourless is observed together with the complete suppression of both characteristic absorption bands of the diarylmethylium tetrafluoroborate salt I (red line in Fig. 2a) and the appearance of a new band in the UV-B region (black line in Fig. 2a). This fact, as pointed out in the reaction optimisation, made us suspect that the catalytic species in this transformation might not be the diarylmethylium salt itself. To better understand the interactions existing between I and the sulfinate 2a, we performed 1H-NMR analysis of a 1/1 mixture of I/2a in CD2Cl2. Ten equivalents of D2O were added to improve sulfinate solubility. As displayed in Fig. 2c, the singlet centred at 9.01 ppm, pertinent to the methenyl (C10–H) between the naphthyl and indolyl moieties of I, disappeared. Moreover, the C2–CH3 was shielded and the corresponding singlet was shifted by 0.88 ppm. The signals at 3.55, 6.82, and 8.01 ppm were subjected not only to a shift but also to a broadening indicating a direct interaction between I and sulfinate 2a.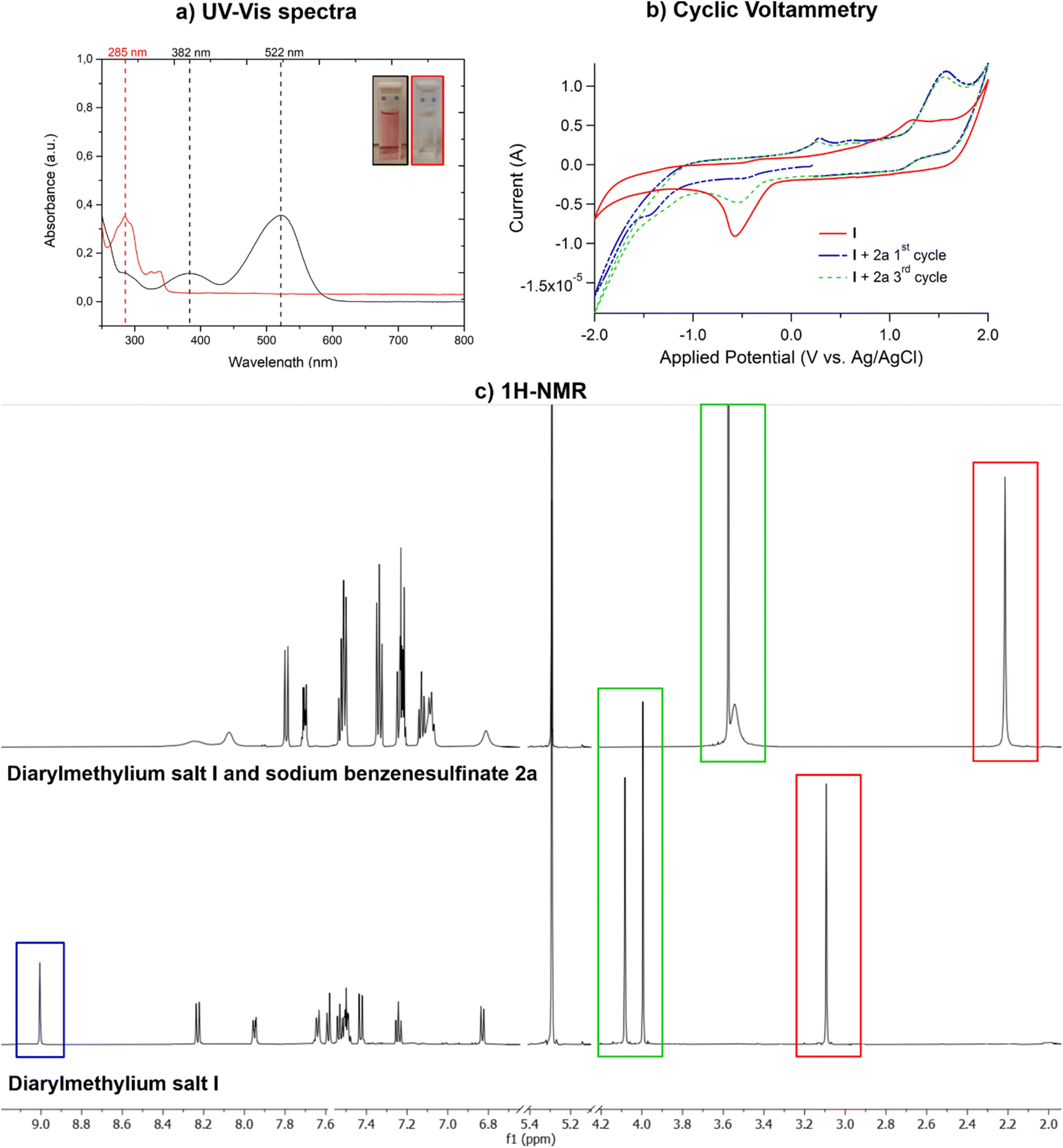 | ||
| Fig. 2 (a) UV-vis spectra of diarylcarbenium salt I (20 mM) in CH2Cl2 before (black line) and after (red line) the addition of sodium benzenesulfinate 2a. (b) CV recorded in CH2Cl2 at 200 mV s−1 of I with and without an excess of 2a. (c) Comparison between the 1H-NMR (600 MHz) spectra of salt I alone (bottom) and in the presence of 2a (top) in CD2Cl2 with 10 equivalents of D2O. | ||
Cyclic voltammetry (CV) demonstrated that the diarylmethylium salts have an irreversible reduction peak at −0.5 V vs. Ag/AgCl as the most distinguishable redox feature (Fig. 2b and ESI†). In the case of I, this characteristic peak disappeared in the first cycle in the presence of sulfinate 2a (Fig. 2b). Moreover, when subjected to repeated cycles, its current was significantly reduced (ESI†). In contrast, this characteristic peak completely disappeared for the diarylmethylium salts not showing a good catalytic activity in the hydrosulfonylation. (see the ESI†). The anodic current peaks observed in the presence of sulfinate 2a were assigned to the sulfinate moiety (Fig. 2b), because very similar features were observed when sulfinate 2a alone was dissolved in methanol (Fig. S26 in the ESI†). Conversely, sulfinate 2a did not give redox features in CH2Cl2, due to its scarce solubility. Therefore, we hypothesized that sulfinate 2a and diarylmethylium salts might form an adduct increasing the solubility of sulfinate in CH2Cl2. Moreover, the adduct should have a charge-transfer nature, as witnessed by the changes in the current of the cathodic peak and in both current and potential of the anodic peaks assigned to the sulfinate moiety (Fig. 2b and ESI†). Therefore, to clarify the interactions between the catalyst and the sulfinate, quantum chemical calculations were carried out.
Computational study
Computational studies were performed on pent-1-ene 1d as the model alkene, tetrafluoroborate I (hereafter, indolynium I+) as the catalyst and acetic acid as the co-reactant and co-catalyst. The analysis of the different configurations and conformations (Tables S-1a/b†) and the electronic structure (group charges and LUMO, Fig. S-1†) of the catalyst I are reported in the ESI-Computational Data (ESI-CD†). In discrete agreement with the experimental findings, the first active electronic transition for catalyst I was calculated using time-dependent density functional theory (TD-DFT) at 484–494 nm (versus the experimental 522 nm, see Fig. 2a). It corresponds to the transition from the HOMO, mainly localized on the naphthyl moiety, to the LUMO orbital, mainly situated on the indolynium unit (see Fig. S-2 in the ESI-CD†). For the mixture catalyst I/sodium benzenesulfinate 2a, calculations predicted the formation of two intermediates in equilibrium between themselves (ESI-CD, Table S-2a†). The most stable is an S–C adduct AD, where the sulphur is directly bound to the methenyl carbon C10–H (Fig. 3, left). | ||
| Fig. 3 Structures of the S–C bound adducts AD (left) and the complex A between sodium benzenesulfinate 2a and the catalyst I (right). Color code: black = carbon, white = hydrogen, blue = nitrogen, red = oxygen, yellow = sulfur. The distances are in Ångström. | ||
This adduct is 9.8 kcal mol−1 more stable than the separated species in terms of free energy. In agreement with the experimental findings, the calculated first electronic transition at 358 nm for adduct AD matched the lack of absorbance in the visible region observed when I and 2a are mixed together (see Fig. 2a for the experimental UV-vis spectrum). The electronic structure of the first excited state (Fig. S-3, ESI-CD†) is mainly described by a transition that involves π and π* orbitals localized on the three aromatic moieties. The contributions of the SO2 moiety and of the σCS* (essential for the dissociation of this bond) are negligible. This, along with the fact that AD does not absorb in the visible, should exclude the creation of the radical couple by photodissociation of the C–S bound adduct. Moreover, the calculated 1H-NMR isotropic magnetic shielding tensors were qualitatively coherent with the experimental findings (Fig. 2c) with a calculated shift for most hydrogen atoms between −1.5 and +0.5 ppm going from I to the S–C adduct AD. The highest upshift of +3.2 ppm was calculated for the C10–H.
As shown in Fig. 3 right, the second species is the complex A, an ion couple formed between 2a and I characterized by a hydrogen bond between the C10–H and one of the oxygens of the sulfinate 2a. This complex is located 7.4 kcal mol−1 below the reactants, but only 2.5 kcal mol−1 above the S–C adduct AD. Therefore, according to our calculations the covalent adduct AD should be the prevailing species, while very small concentrations (ca. 2%) of complex A are present in solution. A similar case was, recently, reported by the Gaunt group.47 Despite the low amount of complex A, it is the key reactive species. Indeed, as indicated by the TD-DFT calculations, its first excited state S1 showed a charge-transfer character that can prelude to the generation of a couple of radicals, i.e. the phenyl sulfonyl radical 2a˙ and the indolyl radical I˙. In fact, this S1 state corresponds to an electronic transition from the HOMO localized on the anionic SO2− moiety to the LUMO localized on the indolinium I+ (see Fig. S-4a in the ESI-CD†). To simplify the model (and to solve the problems of convergence in the optimisation of the first excited state with the TD-DFT calculations, data in Table S-2b, ESI-CD†), we optimised the structures of the complex between the indolyl cation I+ and the sodium benzenesulfinate 2a− without their counter-ions (i.e. Na+ and BF4−). The structure of this new complex, A′ (Fig. S-4b in the ESI-CD†) is similar to that with the counterions and its first excited state also showed the same charge-transfer character (i.e. a transition from the HOMO localized on the anionic SO2− moiety to the LUMO localized on the indolinium I+, see Fig. S-4c in the ESI-CD†). The optimisation of the structure of the first excited state of complex A′ leads to S1-A′* (Fig. 4, left) which is located, in terms of electronic energy (ΔETZ, free energies are not available at the TD-DFT level) 32.7 kcal mol−1 above A, but only 16.8 kcal mol−1 with respect to the separated ions 2a− and I+. Also S1-A* shows an electronic structure corresponding to the couple of radicals formed by the phenylsulfonyl 2a and the indolyl radical I˙. This can also be deduced from the differential density map reported in Fig. 4, right and from the group charges (Fig. S-5 in the ESI-CD†). Therefore, we hypothesized that, upon irradiation, the carbocation I+ is able to reduce itself to the radical I˙, thus oxidizing 2a− to the sulfonyl radical 2a˙ once the complex A− is formed (Scheme 2). The formation of 2a˙ was also confirmed by a trapping experiment with BHT (Fig. 1b). We want to underline that, as experimentally found, this redox reaction is not spontaneous but it requires light to take place.
 | ||
| Fig. 4 Structure of the first excited state S1-A′* of the complex A′ between sodium benzenesulfinate 2a− and indolynium I+ (left) and differential density map (right, the red areas correspond to a reduction in the electronic density when going from S0 to S1, and the blue areas correspond to an increase in the electronic density). Color code: black = carbon, white = hydrogen, blue = nitrogen, red = oxygen, yellow = sulfur. The distances are in Ångström. | ||
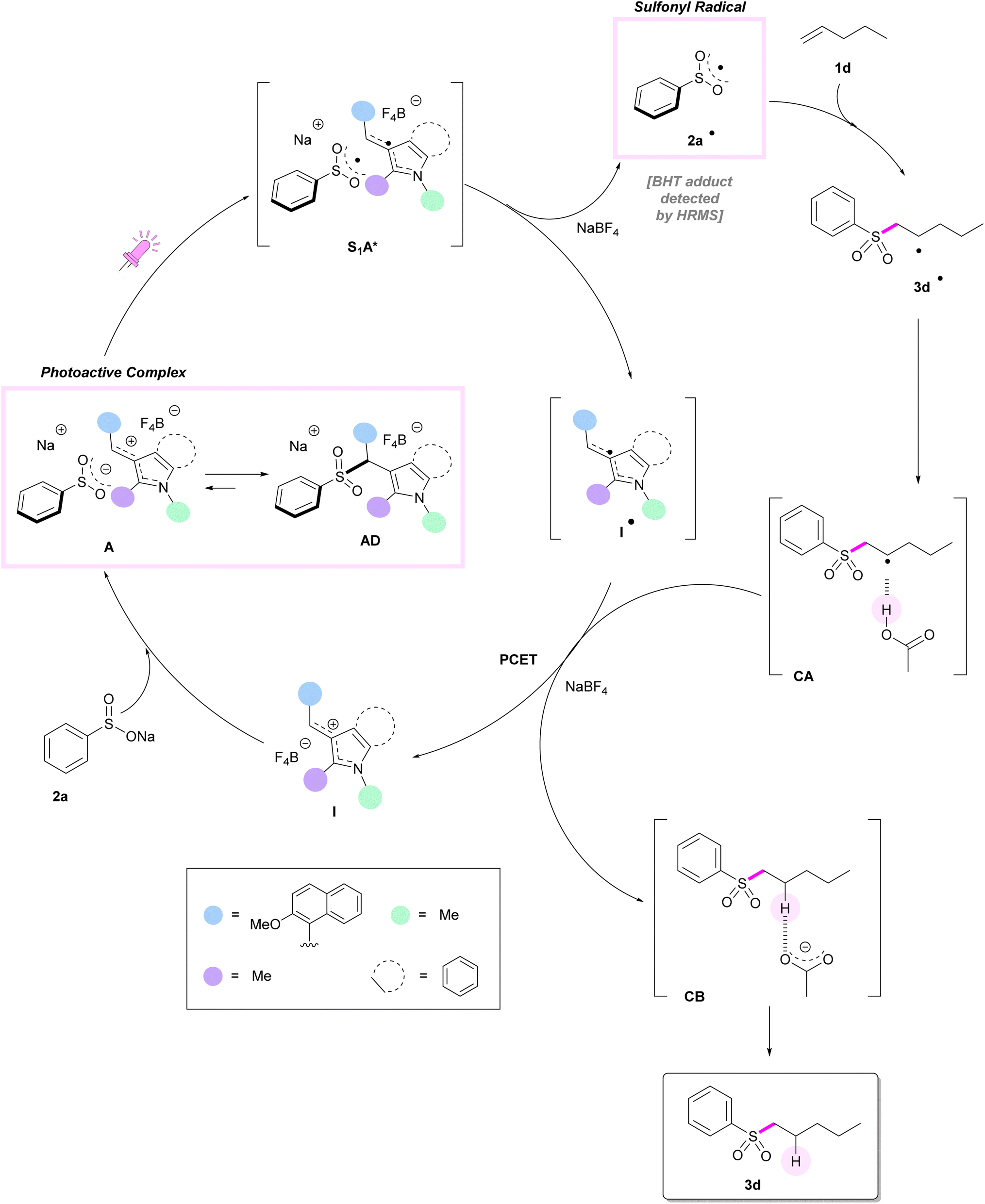 | ||
| Scheme 2 Proposed full reaction mechanism. | ||
S1-A′* can decay to the original A′ or to its corresponding triplet states T1-A′, located 16.1 kcal mol−1 above 2a− and I+ in terms of free energy (but 10.1 kcal mol−1 below S1-A′* in terms of electronic energy). Both S1-A′* and T1-A′ can dissociate into the separated radicals 2a˙ and I˙ which are located 15.0 kcal mol−1 above the ions in terms of free energy, but 1.1 kcal mol−1 below T1-A′ and c.a. 11.2 kcal mol−1 below S1-A′* (assuming the same thermal correction as for the triplet state which is structurally and electronically similar to the first excited singlet state).
As described in Scheme 2, once generated, the radical 2a˙ can add to the alkene 1d to yield intermediate 3d˙ which is a secondary radical and the precursor of the product. Its formation is quite fast (ΔG‡ = 10.8 kcal mol−1, k = 1.8 106 M−1 s−1), but thermodynamically slightly disfavoured (ΔG = 1.3 kcal mol−1). The reduction of the radical intermediate 3d˙ by the reduced indolyl radical I˙ followed by protonation by acetic acid yielding the final product is thermodynamically favoured (ΔG = −19.1 kcal mol−1), but the process seems to be more complicated. As itself, the reduction of the radical 3d˙ by the oxidation of the radical I˙ to regenerate the carbocation catalyst I+ is thermodynamically disfavoured (the estimated value by single point calculation is ΔETZ = 32.7 kcal mol−1). Moreover, the anionic state of 3d˙ is not stable and spontaneously breaks into the starting reactants. However, if one considers the structure of CA between radical 3d˙ and acetic acid and its re-optimised structure after reduction (i.e.CA as an anion), a proton transfer spontaneously takes place yielding CB between the final product 3d and acetate which is located 23.1 kcal mol−1 below the starting CA in terms of energy (ΔETZ, see Fig. S-6 in the ESI-CD†). The final CB is 20.8 kcal mol−1 below CA in terms of free energy. Therefore, in our hypothesis, the reduction and proton transfer can take place in a sort of proton-coupled-electron-transfer, PCET.48–51 The favourable energetics related to the proton transfer step, in fact, compensates the unfavourable energetics of the electron transfer event. In particular, this case could be classified as a reductive multiple-site PCET,52 where the interaction between the carbon radical and the acid could be favoured by the high concentration of the latter in lieu of the molecular structure of the reactant. In our case, the employment of higher than stoichiometric concentrations of acid and the result of the incorporation of deuterium are coherent with this PCET mechanism. Therefore, the acid can induce the reduction of the radical intermediate 2a˙ yielding the thermodynamically stable products. A final counter-ion recombination yields product 3d, sodium acetate and the regenerated catalyst I (ΔG = −22.2 kcal mol−1). The whole reaction is only slightly thermodynamically favoured (ΔG = −1.7 kcal mol−1 in CH2Cl2 and −6.0 kcal mol−1 in water). The details of the computational method are reported in ESI-CD (p. S2†).
Scope of the reaction
With the optimised reaction conditions in hand, we tested this new catalytic protocol with several alkenes demonstrating its wide applicability and its tolerance towards a plethora of functional groups with different electronic properties. The results are reported in Table 2. Reactions were carried out both on 0.5 and 1 mmol scales with no meaningful differences in the outcome. For instance, olefins with different chain lengths were tested and the best results were observed with hexadec-1-ene 1a and oct-1-ene 1c, which both afforded the corresponding sulfones 3a and 3c in 80% yields. Slightly lower yields were observed with dodec-1-ene 1b (3b, 66%), pent-1-ene 1d (3d, 57%) and with cyclohexene 1f (3f, 50%). Remarkably, ethylene 1e could also be employed at atmospheric pressure, affording sulfone 3e in a 40% yield. As shown in Table 2, our methodology was successfully applied to many functionalised olefins producing a library of substituted sulfones. Leaving groups and protic groups (respectively –Br and –OH) were tolerated, as demonstrated in the case of 4-bromobut-1-ene 1g which produced the corresponding sulfone 3g in a very good yield, both when 0.5 and 1 mmol of 1g were used (69 and 79%, respectively). The same was observed with but-3-en-1-ol (1h) and hex-5-en-1-ol (1i), whose corresponding sulfones 3h and 3i were obtained in 78 and 90% yields, respectively. Interestingly, sulfone 3i was achieved in a higher yield than that reported by Yu25 applying iridium photocatalysis (90% versus 69% yield). An ether functional group was also tolerated, with 2-allyloxyethanol 1j delivering the corresponding sulfone 3j in a 74% yield. Ketones and epoxy rings were easily introduced onto the sulfone product as shown in the case of hex-1-en-2-one 1k and of 5,6-epoxyhex-1-ene 1l (52% yield for 3k, quantitative yield for 3l). Our strategy can be also applied to α,β-unsaturated enals or enones, but it did not prove to be advantageous compared to the polar protocol affording the corresponding β-sulfonyl-ketones or aldehydes in lower yields. Considering alkenes functionalised at the allylic position, the formation of the sulfone product can be directly correlated to the leaving-group nature of the substituent. For allyl bromide, only (allylsulfonyl)benzene, which was derived from nucleophilic displacement, was produced. However, allyl chloride 1m, produced the corresponding ((3-chloropropyl)sulfonyl)benzene 3m in a 35% yield, because of the worse leaving ability of chloride than that of bromide, but still in the presence of 18% of (allylsulfonyl)benzene. The reaction was also successfully accomplished with allyl alcohol 1n, with the corresponding phenylsulfonylpropan-1-ol 3n produced in 63% yield. Interestingly, the nucleophilic displacement was totally suppressed with allylboronic acid pinacol ester 1o and allyltrimethylsilane 1p. Sulfone 3o was obtained in an excellent 96% yield. This is a remarkable result considering that allylboronic esters and allyl silanes can be exploited as allylation reagents where boronic ester or silane moieties get lost or 1,2-boron migration is observed.53–56 Considering the high synthetic value of compound 3o as a possible substrate for a Suzuki coupling, we scaled-up its synthesis to 15 mmol with a yield of 73% (see the ESI† for pictures of the reaction work flow for large scale sulfonylation). The use of 1p was also successful with sulfone 3p produced in 82% yield. Taking into account polymerization as a side reaction associated with acrylonitrile 1q, 3-(phenylsulfonyl)propanenitrile 3q was obtained in a satisfactory 50% yield. However, when N,N-dimethylacrylamide 1r was used as the reagent, a drop in yield to 15% for 3r was observed. Moreover, carboxylic acid (1s–t) or ester (1u) moieties were very well tolerated, with the isolation of the corresponding sulfones 3s, 3t and 3u, respectively in 92, 67 and 62% yields. Masked amine functionality could be easily introduced from 4-phtalimidobut-1-ene 1v (71% for 3v). A similar result was observed when a tosylamine was incorporated in the starting alkene with the product 3w produced in 70% yield. Styrenes were also tested, but no sulfonylation product was obtained. Finally, the hydrosulfonylation protocol was validated on natural and biologically active molecules. (5R)-(+)-Pulegone derivative 1x produced the corresponding product 3x in a 43% yield as a diastereomeric mixture in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.3 ratio. Remarkably, (4R)-(−)-limonene 1y afforded the product 3y with complete selectivity to the terminal double bond. Hydrosulfonylated analogues of (S)-(+)-Ibuprofen and cholesterol derivatives were isolated in good yields (60% for 3za and 74% for 3zb, respectively). In order to obtain the full conversion of 1zb, the reaction time was increased to 46 h (35% yield for 3zb after 23 h).
:1.3 ratio. Remarkably, (4R)-(−)-limonene 1y afforded the product 3y with complete selectivity to the terminal double bond. Hydrosulfonylated analogues of (S)-(+)-Ibuprofen and cholesterol derivatives were isolated in good yields (60% for 3za and 74% for 3zb, respectively). In order to obtain the full conversion of 1zb, the reaction time was increased to 46 h (35% yield for 3zb after 23 h).
| a Reaction conditions: alkene 1a–z (0.5 mmol), sodium benzenesulfinate 2a (1.6 eq.), I (1 mol%), in CH2Cl2 (2.5 mL, 0.2 M), CH3COOH (4.5 eq.), H2O (10 eq.), 40W Kessil purple LED lamp (390 nm), 23 h, room temperature. b Yield determined on the isolated product. c Reaction carried out on 1 mmol of 1c–e, 1g, 1i–v and 1y in 5 mL of CH2Cl2. d Reaction scaled up to 15 mmol of 1o. e Reaction time 46 h for 1zb. |
|---|

|
The scope of the sulfinate was then studied, and sodium 4-methylbenzenesulfinate 2b was first applied affording the products 4a–f in good to excellent yields, comparable to those obtained with sodium benzenesulfinate 2a. Sulfones 4a and 4b were obtained by reaction with hexadec-1-ene 1a and oct-1-ene 1c in 71 and 78% yield, respectively. Also in the case of (4R)-(−)-limonene 1w the outcome was comparable to that of sulfinate 2a (57% vs. 55%) with a complete selectivity toward the less hindered double bond. Actually, simple alkenes successfully reacted with all the substituted sulfinates. Hexadec-1-ene 1a, for example, afforded sulfone 4g in 84% yield after reaction with sodium 4-chlorobenzenesulfinate 1c. The shorter alkene 1c gave sulfone 4i in nearly quantitative yield (97%). In the case of sodium 4-acetylbenzenesulfinate 1d and the sterically hindered mesityl sulfinate 1e a drop in yield was obtained (4m, 20% and 4o, 15%). Sulfone 4f, derived from 11-bromoundec-1-ene 1aa and p-tolylsulfinate 2b, was produced in 88% yield thus confirming the compatibility of this protocol with alkyl bromides. But-3-enol 1h successfully reacted with 2b (4d, 56%), with a lower efficiency than 2a. The hydrosulfonylation of allylpinacolborane 1n was successfully performed with several sulfinates (see Table 3, products 4e, 79% and 4j 84% yield). Anyway, a marked decrease in yield was observed in the case of sulfinates 2d and 2e (4n and 4p, 23 and 25% yield, respectively), consistent with the results obtained with the same sulfinates with other alkenes, as previously described. Sulfone 4k was isolated in 50% yield by the reaction of allyltrimethylsilane 1o and p-Cl-sulfinate 2c thus confirming a higher efficiency of 2a with respect to other derivatives. Interestingly product 4l bearing a ketone functionality, although in low yield, was obtained from 3-penten-2-ol 1ab after a rearrangement.
| a Reaction conditions: alkene 1 (0.5 mmol), sodium aryl sulfinates 2b–e (1.6 eq.), I (1 mol%), in CH2Cl2 (2.5 mL, 0.2 M), CH3COOH (4.5 eq.), H2O (10 eq.), 40W Kessil purple LED lamp (390 nm), 23 h, room temperature. b Yield determined on the isolated product. c Reaction carried out on 1 mmol of alkenes 1b–c, 1e–g, 1j and 1o in 5 mL of CH2Cl2. |
|---|

|
Conclusions
In summary, we have developed a general light mediated hydrosulfonylation of unactivated alkenes using substituted aryl sulfinates in the presence of diarylmethylium tetrafluoroborates. These salts, which are cheap, stable under air and easily prepared on a multigram scale, were employed for the first time in a photoredox process with the same loading of iridium-based photocatalysts. This methodology, which is operationally simple and do not require anhydrous conditions, allowed the access to more than 40 functionalised sulfones with a remarkable functional group tolerance and scalability up to 15 mmol. Natural products and biologically active molecules were hydrosulfonylated under mild reaction conditions. Experimental and computational mechanistic studies revealed that the key sulfonyl radical can be formed through an innovative mechanism involving a photoactive adduct derived from the diarylmethylium salt and the sulfinate upon interaction with light. A PCET process is then exploited to restore the catalytic species.Data availability
The data, both experimental and computational, that supports the findings of this study are available in the ESI of this article.†Author contributions
All authors have given approval to the final version of the manuscript. The manuscript was written through contributions of all authors. Polyssena Renzi (conceptualization, methodology, investigation, visualization, writing – original draft, writing review and editing), Emanuele Azzi, Sylvain Ascensio, Stefano Parisotto, Fabrizio Sordello, and Francesco Pellegrino (investigation, writing review and editing), Giovanni Ghigo (investigation, software, writing – original draft, writing review and editing), and Annamaria Deagostino (conceptualization, funding acquisition, project administration, supervision, writing – original draft, writing – review & editing).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is dedicated to Dr Margherita Barbero on the occasion of her retirement. This work was supported by MIUR (Ministero dell’Istruzione, dell’Università e della Ricerca).References
- M. Feng, B. Tang, S. H. Liang and X. Jiang, Sulfur Containing Scaffolds in Drugs: Synthesis and Application in Medicinal Chemistry, Curr. Top. Med. Chem., 2016, 16, 1200–1216 CrossRef CAS.
- K. A. Scott and J. T. Njardarson, Analysis of US FDA-Approved Drugs Containing Sulfur Atoms, Curr. Top. Med. Chem., 2018, 376, 5 CrossRef.
- D. Joseph, M. A. Idris, J. Chen and S. Lee, Recent Advances in the Catalytic Synthesis of Arylsulfonyl Compounds, ACS Catal., 2021, 11, 4169–4204 CrossRef CAS.
- S. Liang, K. Hofman, M. Friedrich, J. Keller and G. Manolikakes, Recent Progress and Emerging Technologies towards a Sustainable Synthesis of Sulfones, ChemSusChem, 2021, 14, 4878–4902 CrossRef CAS.
- B. M. Trost and D. P. Curran, Chemoselective oxidation of sulfides to sulfones with potassium hydrogen persulfate, Tetrahedron Lett., 1981, 22, 1287–1290 CrossRef CAS.
- W. Su, An efficient method for the oxidation of sulfides to sulfones, Tetrahedron Lett., 1994, 35, 4955–4958 CrossRef CAS.
- S. Hussain, S. K. Bharadwaj, R. Pandey and M. K. Chaudhuri, Borax-Catalyzed and pH-Controlled Selective Oxidation of Organic Sulfides by H2O2: An Environmentally Clean Protocol, Eur. J. Org. Chem., 2009, 2009, 3319–3322 CrossRef.
- K. Bahrami, M. M. Khodaei and M. Sheikh Arabi, TAPC-Promoted Oxidation of Sulfides and Deoxygenation of Sulfoxides, J. Org. Chem., 2010, 75, 6208–6213 CrossRef CAS.
- S. Ye, G. Qiu and J. Wu, Inorganic sulfites as the sulfur dioxide surrogates in sulfonylation reactions, Chem. Commun., 2019, 55, 1013–1019 RSC.
- G. Qiu, K. Zhou and J. Wu, Recent advances in the sulfonylation of C–H bonds with the insertion of sulfur dioxide, Chem. Commun., 2018, 54, 12561–12569 RSC.
- S. Ye, M. Yang and J. Wu, Recent advances in sulfonylation reactions using potassium/sodium metabisulfite, Chem. Commun., 2020, 56, 4145–4155 RSC.
- A. Lanfranco, R. Moro, E. Azzi, A. Deagostino and P. Renzi, Unconventional approaches for the introduction of sulfur-based functional groups, Org. Biomol. Chem., 2021, 19, 6926–6957 RSC.
- L. Wu, L. Peng, Z. Hu, Y. Jiao and Z. Tang, Recent Advances of Sulfonylation Reactions in Water, Curr. Org. Synth., 2020, 17, 271–281 CrossRef CAS.
- E. Azzi, A. Lanfranco, R. Moro, A. Deagostino and P. Renzi, Visible light as the key for the formation of carbon-sulfur bonds in sulfones, thioethers, and sulfonamides: An update, Synthesis, 2021, 53, 3440–3468 CrossRef CAS.
- Z. Lu, M. Shang and H. Lu, Organic Sulfinic Acids and Salts in Visible Light-Induced Reactions, Synthesis, 2022, 54(5), 1231–1249 CrossRef CAS.
- D.-Q. Dong, Q.-Q. Han, S.-H. Yang, J.-C. Song, N. Li, Z.-L. Wang and X.-M. Xu, Recent Progress in Sulfonylation via Radical Reaction with Sodium Sulfinates, Sulfinic Acids, Sulfonyl Chlorides or Sulfonyl Hydrazides, ChemistrySelect, 2020, 5, 13103–13134 CrossRef CAS.
- O. M. Mulina, A. I. Ilovaisky, V. D. Parshin and A. O. Terent'ev, Oxidative Sulfonylation of Multiple Carbon-Carbon bonds with Sulfonyl Hydrazides, Sulfinic Acids and their Salts, Adv. Synth.Catal., 2020, 362, 4579–4654 CrossRef CAS.
- X. Ye, X. Wu, S.-r. Guo, D. Huang and X. Sun, Recent advances of sodium sulfinates in radical reactions, Tetrahedron Lett., 2021, 81, 153368 CrossRef CAS.
- B. Ni, B. Zhang, J. Han, B. Peng, Y. Shan and T. Niu, Heterogeneous carbon nitrides photocatalysis multicomponent hydrosulfonylation of alkynes to access β-keto sulfones with the insertion of sulfur dioxide in aerobic aqueous medium, Org. Lett., 2020, 22, 670–674 CrossRef CAS.
- X. Du, J.-S. Zhen, X.-H. Xu, H. Yuan, Y.-H. Li, Y. Zheng, C. Xue and Y. Luo, Hydrosulfonylation of Alkenes with Sulfonyl Imines via Ir/Cu Dual Photoredox Catalysis, Org. Lett., 2022, 24, 3944–3949 CrossRef CAS.
- A. Hossain, S. Engl, E. Lutsker and O. Reiser, Visible-Light-Mediated Regioselective Chlorosulfonylation of Alkenes and Alkynes: Introducing the Cu(II) Complex [Cu(dap)Cl2] to Photochemical ATRA Reactions, ACS Catal., 2019, 9, 1103–1109 CrossRef CAS.
- M. Alkan-Zambada and X. Hu, Cu-Catalyzed Photoredox Chlorosulfonation of Alkenes and Alkynes, J. Org. Chem., 2019, 84, 4525–4533 CrossRef CAS.
- S. K. Pagire, S. Paria and O. Reiser, Synthesis of β-Hydroxysulfones from Sulfonyl Chlorides and Alkenes Utilizing Visible Light Photocatalytic Sequences, Org. Lett., 2016, 18, 2106–2109 CrossRef CAS.
- X.-J. Tang and W. R. Dolbier Jr, Efficient Cu-catalyzed Atom Transfer Radical Addition Reactions of Fluoroalkylsulfonyl Chlorides with Electron-deficient Alkenes Induced by Visible Light, Angew. Chem., Int. Ed., 2015, 54, 4246–4249 CrossRef CAS.
- J. J. Wang and W. Yu, Hydrosulfonylation of Unactivated Alkenes by Visible Light Photoredox Catalysis, Org. Lett., 2019, 21, 9236–9240 CrossRef CAS.
- Y. Zheng, Y. You, Q. Shen, J. Zhang, L. Liu and X.-H. Duan, Visible-light-induced anti-Markovnikov hydrosulfonation of styrene derivatives, Org. Chem. Front., 2020, 7, 2069–2074 RSC.
- Y. Chen, N. McNamara, O. May, T. Pillaiyar, D. C. Blakemore and S. V. Ley, Photoredox Generation of Sulfonyl Radicals and Coupling with Electron Deficient Olefins, Org. Lett., 2020, 22, 5746–5748 CrossRef CAS.
- S. M. Hell, C. F. Meyer, A. Misale, J. B. I. Sap, K. E. Christensen, M. C. Willis, A. A. Trabanco and V. Gouverneur, Hydrosulfonylation of Alkenes with Sulfonyl Chlorides under Visible Light Activation, Angew. Chem., Int. Ed., 2020, 59, 11620–11626 CrossRef CAS.
- T. Föll, J. Rehbein and O. Reiser, Ir(ppy)3-Catalyzed, Visible-Light-Mediated Reaction of α-Chloro Cinnamates with Enol Acetates: An Apparent Halogen Paradox, Org. Lett., 2018, 20, 5794–5798 CrossRef.
- Britannica, The Editors of Encyclopaedia, iridium, Encyclopedia Britannica, 29 Jan. 2021, https://www.britannica.com/science/iridium, accessed 20 March 2022 Search PubMed.
- A. Joshi-Pangu, F. Lévesque, H. G. Roth, S. F. Oliver, L.-C. Campeau, D. Nicewicz and D. A. DiRocco, Acridinium-Based Photocatalysts: A Sustainable Option in Photoredox Catalysis, J. Org. Chem., 2016, 81, 7244–7249 CrossRef CAS.
- J. Cervantes-González, D. A. Vosburg, S. E. Mora-Rodriguez, M. A. Vázquez, L. G. Zepeda, C. Villegas Gómez and S. Lagunas-Rivera, Anthraquinones: Versatile Organic Photocatalysts, ChemCatChem, 2020, 12, 3811–3827 CrossRef.
- Y. Lee and M. S. Kwon, Emerging Organic Photoredox Catalysts for Organic Transformations, Eur. J. Org. Chem., 2020, 2020, 6028–6043 CrossRef CAS.
- V. Srivastava and P. P. Singh, Eosin y catalysed photoredox synthesis: A review, RSC Adv., 2017, 7, 31377–31392 RSC.
- N. A. Romero and D. A. Nicewicz, Organic Photoredox Catalysis, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS.
- C. Pezzetta, A. Folli, O. Matuszewska, D. Murphy, R. W. M. Davidson and D. Bonifazi, peri-Xanthenoxanthene (PXX): a Versatile Organic Photocatalyst in Organic Synthesis, Adv. Synth. Catal., 2021, 363, 4740–4753 CrossRef CAS.
- P. Renzi, E. Azzi, E. Bessone, G. Ghigo, S. Parisotto, F. Pellegrino and A. Deagostino, Blue light enhanced Heck arylation at room temperature applied to allenes, Org. Chem. Front., 2022, 9, 906–916 RSC.
- E. Azzi, G. Ghigo, S. Parisotto, F. Pellegrino, E. Priola, P. Renzi and A. Deagostino, Visible Light Mediated Photocatalytic N-Radical Cascade Reactivity of γ,δ-Unsaturated N-Arylsulfonylhydrazones: A General Approach to Structurally Diverse Tetrahydropyridazines, J. Org. Chem., 2021, 86, 3300–3323 CrossRef CAS.
- S. Parisotto, G. Garreffa, C. Canepa, E. Diana, F. Pellegrino, E. Priola, C. Prandi, V. Maurino and A. Deagostino, Visible-Light-Driven Photocatalytic Transformation of α,β-Unsaturated-N-Tosylhydrazones: A Novel Route to Allylic Sulfones, ChemPhotoChem, 2017, 1, 56–59 CrossRef CAS.
- N. Armenise, S. Dughera, A. Gualandi, L. Mengozzi, M. Barbero and P. G. Cozzi, Organocatalyzed Asymmetric Alkylation of Stable Aryl or Heteroaryl(3-indolyl)methylium o-Benzenedisulfonimides, Asian J. Org. Chem., 2015, 4, 337–345 CrossRef CAS.
- M. Barbero, R. Buscaino, S. Cadamuro, S. Dughera, A. Gualandi, D. Marabello and P. G. Cozzi, Synthesis of Bench-Stable Diarylmethylium Tetrafluoroborates, J. Org. Chem., 2015, 80, 4791–4796 CrossRef CAS.
- M. Barbero, S. Cadamuro, F. Cauda, S. Dughera, G. Gervasio and P. Venturello, Preparation and Characterization of Aryl or Heteroaryl(3-indolyl)methylium o-Benzenedisulfonimides, J. Org. Chem., 2012, 77, 4278–4287 CrossRef CAS.
- M. Barbero, S. Cadamuro, S. Dughera, G. Ghigo, D. Marabello and P. Morgante, Efficient alkylation of cyclic silyl enol ethers by diarylmethylium salts, Tetrahedron Lett., 2016, 57, 4758–4762 CrossRef CAS.
- R. J. Mayer, N. Hampel, P. Mayer, A. R. Ofial and H. Mayr, Synthesis, Structure, and Properties of Amino-Substituted Benzhydrylium Ions – A Link between Ordinary Carbocations and Neutral Electrophiles, Eur. J. Org. Chem., 2019, 2019, 412–421 CrossRef CAS.
- R. J. Mayer, A. R. Ofial, H. Mayr and C. Y. Legault, Lewis Acidity Scale of Diaryliodonium Ions toward Oxygen, Nitrogen, and Halogen Lewis Bases, J. Am. Chem. Soc., 2020, 142, 5221–5233 CrossRef CAS.
- On a relative molar scale, diarylcarbenium salt I is more than three hundred times cheaper than [Ir(dF(CF3)ppy)2(dtbbpy)]PF6. For the calculation of diarylcarbenium salt I cost and comparison with prices of iridium catalysts see the ESI†.
- K. Kohara, A. Trowbridge, M. A. Smith and M. J. Gaunt, Thiol-Mediated α-Amino Radical Formation via Visible-Light-Activated Ion-Pair Charge-Transfer Complexes, J. Am. Chem. Soc., 2021, 143, 19268–19274 CrossRef CAS.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Proton-Coupled Electron Transfer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS.
- R. Tyburski, T. Liu, S. D. Glover and L. Hammarström, Proton-Coupled Electron Transfer Guidelines, Fair and Square, J. Am. Chem. Soc., 2021, 143, 560–576 CrossRef CAS.
- R. G. Agarwal, S. C. Coste, B. D. Groff, A. M. Heuer, H. Noh, G. A. Parada, C. F. Wise, E. M. Nichols, J. J. Warren and J. M. Mayer, Free Energies of Proton-Coupled Electron Transfer Reagents and Their Applications, Chem. Rev., 2022, 122, 1–49 CrossRef CAS.
- P. R. D. Murray, J. H. Cox, N. D. Chiappini, C. B. Roos, E. A. McLoughlin, B. G. Hejna, S. T. Nguyen, H. H. Ripberger, J. M. Ganley, E. Tsui, N. Y. Shin, B. Koronkiewicz, G. Qiu and R. R. Knowles, Photochemical and Electrochemical Applications of Proton-Coupled Electron Transfer in Organic Synthesis, Chem. Rev., 2022, 122, 2017–2291 CrossRef CAS.
- T. F. Markle, J. W. Darcy and J. M. Mayer, A new strategy to efficiently cleave and form C–H bonds using proton-coupled electron transfer, Sci. Adv., 2018, 4, eaat5776 CrossRef.
- R. Wada, K. Oisaki, M. Kanai and M. Shibasaki, Catalytic Enantioselective Allylboration of Ketones, J. Am. Chem. Soc., 2004, 126, 8910–8911 CrossRef CAS.
- S. E. Denmark and J. Fu, Catalytic Enantioselective Addition of Allylic Organometallic Reagents to Aldehydes and Ketones, Chem. Rev., 2003, 103, 2763–2794 CrossRef CAS.
- M. Berger, D. Carboni and P. Melchiorre, Photochemical Organocatalytic Regio- and Enantioselective Conjugate Addition of Allyl Groups to Enals, Angew. Chem., Int. Ed., 2021, 60, 26373–26377 CrossRef CAS.
- K. Jana and A. Studer, Allylboronic Esters as Acceptors in Radical Addition, Boron 1,2-Migration, and Trapping Cascades, Org. Lett., 2022, 24, 1100–1104 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, materials, and methods, including photographs of the experimental setup and NMR spectra of all compounds (PDF). Details of the computational method, additional discussions and figures, table with absolute and relative energies, pictures of all structures (with imaginary frequencies for the TSs) and relative Cartesian coordinates (PDF). See DOI: https://doi.org/10.1039/d3sc00182b |
| This journal is © The Royal Society of Chemistry 2023 |