Formation of nanostructures by self-assembly of an elastin peptide†
Antonietta
Pepe
,
Maria Rosaria
Armenante
,
Brigida
Bochicchio
and
Antonio Mario
Tamburro
*
Department of Chemistry, University of Basilicata, Via N. Sauro 85, 85100 Potenza, Italy. E-mail: antonio.tamburro@unibas.it; Fax: +39 0971 202223; Tel: +39 0971 202258
First published on 14th October 2008
Abstract
Elastin and elastin-related peptides have great potential in the biomaterial field, because of their peculiar mechanical properties and spontaneous self-assembling behavior. Depending on their sequences and under appropriate experimental conditions, they are able to self-assemble in different fiber morphologies, including amyloid-like fibers. Temperature-triggered self-assembly of a small elastin peptide shows a novel complex aggregation mechanism as revealed by different microscopy techniques. The conformations of the peptide have been investigated in solution and in the aggregated state by different spectroscopic techniques (CD, NMR, FT-IR) and revealed that the conformations adopted by the peptides in water in the prefibrillar state correspond to those populated by other elastin peptides, mainly polyproline II helix (PPII) and random coil. Conversely, the aggregated state shows evidence for antiparallel cross-β structures. Our molecular studies highlight the important role of PPII conformation on the prefibrillar state, putting forward the hypothesis that aggregation takes place through addition of the monomer in the PPII conformation with preformed β-sheet aggregates and/or through direct interaction of PPII helices.
Introduction
Molecular self-assembly offers new opportunities for the fabrication of novel supramolecular structures and advanced materials. The development of such structures is often inspired by biological self-assembling modules, where simple building blocks such as amino acids, nucleic acids and lipids are able to form complex natural systems. Peptide-based nanostructures represent an important way toward the production of ordered self-assembling nanostructures with variegated possible applications.1 The growing interest in protein-inspired bionanotechnology is based on the knowledge of different self-assembling processes involving proteins and peptides. One of the most ubiquitous self-assembly processes in nature is the hierarchical organization of protein monomers into long filament bundles and networks of nanometric dimensions.Extracellular matrix proteins such as elastin and collagen are involved in different self-assembling processes, both producing well defined fibrils and fibers with specific mechanical and supramolecular properties.2 Another class of protein-based fibers are the amyloid fibers, formed by different proteins undergoing misfolding and/or misassembling.3 These aggregation patterns are often associated with pathological states of human diseases [Alzheimer’s disease (AD), diabetes], even though recently different studies point to a functional role of amyloid fibers in nature.4
However, because of the mechanical properties of these fibrillar structures, they have considerable potential as novel biomaterials with applications for nanotechnology.5 Among the proteins able to self-assemble, elastin and elastin-related polypeptides6,7 represent a special group from different points of view: (i) the sequences responsible for their self-assembling as well as for the elastic properties are of reduced size and complexity,8 (ii) important mechanical properties can be tuned by the choice of the sequence building block;9 (iii) several sequences are able to self-assemble into two different aggregation patterns, the classical elastin-like6,10 and the amyloid-type.11 These peculiar features of elastin-related polypeptides render them a special subject of interest, as bionanomaterials with “smart” behavior.12
From a molecular point of view, amyloid fibers are composed of β-strands stacked in an orthogonal way with respect to the fiber axis. The resulting cross-β structures are stabilized by hydrogen bonds, although also other types of interactions, electrostatic and/or hydrophobic, are important. The cross-β structure of the fibers is common to almost all the amyloid fibers, even though the monomeric species are unrelated in terms of sequence, size, hydrophobicity, and secondary structures.
In many cases partial denaturation or misfolding is responsible for the aggregation.13 For example, the proteolysis of APP (Alzheimer precursor protein), determines a transition from the native state to a β-sheet structure in the Aβ peptides, responsible for the amyloid aggregation in Alzheimer’s disease (AD). Experimentally, denaturation and consequently protein aggregation is induced through the addition of chaotropic agents, organic solvents (TFE, HFIP), ionic strength of the solution, or under extreme physical perturbations such as high temperature or pressure values. In several other cases, under appropriate experimental conditions, peptides and proteins—the socalled intrinsically unordered proteins (IUP)—devoid of stable secondary structures, evolve to amyloid fibers. For these samples the aggregation proceeds mainly through a PPII to β-structure transition.14 This is not surprising if we consider that both conformations populate the same upper left Ramachandran map region, thus requiring very small dihedral angle variations for the transition to occur. In addition, possible amyloid fibers directly originating from PPII structured polypeptides, have been suggested.15
With the aim of exploring the possible use of elastin self-assembling peptides for biotechnological applications, we searched for the minimal sequence of elastin responsible for the amyloid-like self-assembly. We have previously reported that the exon30-coded domain of human tropoelastin (EX30 30, GLVGAAGLGGLGVGGLGVPGVGGLG) is able to form amyloid fibers, whose diameter and helical pitch is time-dependent.16,17 The syntheses and self-assembling analysis of different fragments allowed us to identify the smallest amyloidogenic peptide, whose sequence corresponds to the first 17 N-terminal residues of EX30N (EX30N, GLVGAAGLGGLGVGGLG). This peptide contains twice the XGGZG sequence (X, Z = V, L), previously suggested to be responsible for amyloid-formation in elastin-related polypeptides.18 In this report, we will show that this 17 residue fragment of human tropoelastin forms a peculiar aggregation pattern, possessing characteristics of the amyloid fibers, with a great tendency to form nano-films. We determined the conformation of the monomeric species, by CD and NMR spectroscopy, and of the aggregate state, by FT-IR spectroscopy. According to our results also for this peptide a PPII to β-structure transition should be suggested as responsible for amyloid aggregation.
Experimental
Peptide synthesis and purification
The peptide sequence was synthesized by a solid phase methodology using an automatic synthesizer APPLIED BIOSYSTEM model 431 A. Fmoc/DCC/HOBT chemistry was used, starting from 222 mg (0.25 mmol) Wang resin (Nova Biochem, Laufelfingen, Switzerland). The Fmoc-amino acids were purchased from Nova Biochem (Laufelfingen, Switzerland) and from Inbios (Pozzuoli, Italy). The cleavage of the peptide from the resin was achieved by using an aqueous mixture of 95% trifluoroacetic acid. The peptide was lyophilized and purified by reversed-phase HPLC (high performance liquid chromatography) (see ESI†). A binary gradient was used and the solvents were H2O (0.1% TFA) and CH3CN (0.1% TFA). The purity of the peptide was assessed by electrospray mass spectrometry (see ESI†).Turbidimetry experiments
The EX30Npeptide (1.5 mM) was dissolved in water. Turbidimetry was measured at 440 nm as a function of time on a Cary UV50 spectrophotometer equipped with a Peltier temperature controller using quartz cuvettes and reported as TAA (turbidimetry on apparent absorbance) vs. time. Turbidimetry was recorded at room temperature and at 50 °C.Circular dichroism (CD) spectroscopy
CD spectra for the peptide were obtained using a Jasco J-600 Spectropolarimeter at various temperatures and at concentrations of 0.1 mg ml−1 in water by using cells of 0.1 cm. Spectra were acquired in the range 190–250 nm by taking points every 0.1 nm, with a 20 nm min−1 scan rate, an integration time of 2 s, and a 1 nm bandwidth. The data are expressed as [θ], the molar ellipticity in units of ° cm2 dmol−1.Nuclear magnetic resonance (NMR) spectroscopy
All 1H NMR experiments were performed on a Varian Unity INOVA 500 MHz spectrometer equipped with a 5 mm triple-resonance probe and z-axial gradients. The purified peptide was dissolved in 700 µl of H2O–D2O (90 : 10, v/v) or in TFE-d3–H2O (80 : 20), containing 0.1 mM of 3-(trimethyl-silyl)-1-propane sulfonic acid (DSS) as an internal reference standard at 0 ppm. 3 mM peptide solutions were used. One-dimensional spectra were acquired in Fourier mode with quadrature detection and the water signal was suppressed by a 2.5 s presaturation pulse. Two-dimensional TOCSY19 and NOESY20 and ROESY spectra were collected in the phase-sensitive mode using the States method. Typical data were 2048 complex data points, 16 or 32 transients and 256 increments. Relaxation delays were set to 2.5 s and spinlock (MLEV-17) mixing time was 80 ms for TOCSY while a 100–200 ms mixing time was applied to NOESY and ROESY experiments. Shifted sine bell squared weighting and zero filling to 2K × 2K was applied before Fourier transformation. The residual HDO signal was suppressed by double-pulsed field-gradient spin-echo.21Amide proton temperature coefficients were usually measured from 1D 1H NMR spectra recorded in 5 °C increments from 20 °C to 45 °C, while for overlapping resonances a series of TOCSY were recorded in order to measure the amide temperature coefficients. Data were processed with NMRPipe.22 Sparky23 was used for data visualization, assignments, and peak integration.Sequential resonance assignments were made by the approach described by Wüthrich.24
Fourier transform infrared (FT-IR) spectroscopy
The peptide was analyzed by FT-IR as precipitated fibers in KBr pellets (1 mg 100 mg−1). The spectra were recorded on a Jasco FT-IR-460 PLUS using a resolution of 4 cm−1 and then smoothed by using the Savintky–Goolay algorithm. The decomposition of FT-IR spectra was obtained using the peak fitting module implemented in the Origin© Software (Microcalc Inc.) using the the second derivative method. In the curve fitting procedure, the Voigt peak shape has been used for all peaks. The Voigt shape is a combination of the Gaussian and Lorentzian peak shapes and accounts for the broadening present in the FT-IR spectrum.Congo red birefringence assay
The staining solution was prepared by using 80% EtOH, 20% water and a saturating amount of Congo Red and NaCl. The solution was stirred for a few minutes, and filtered. Fifty microlitres of EX30N fiber-containing suspension was deposited onto a glass slide and, after air-drying, Congo red staining solution was added. The excess solution was blotted away, and the sample was allowed to dry at room temperature.25 The stained sample was examined under polarized light with a Nikon ECLIPSE E600 POL microscope equipped with a Nikon Coolpix 4500 4.0 megapixel camera.Transmission electron microscopy (TEM)
The peptide was dissolved in water at room temperature at a concentration of 1.5 mM. The solution was divided into vials that were sealed and kept at 50 °C into a thermostat for times of up to 19 h. At fixed intervals, 10 µl of the incubated solution were placed on formvar and carbon-coated copper grids, negatively stained with a few drops of 1% uranyl acetate in bidistilled water, air dried and observed by a ZEISS EM 10C transmission electron microscopy.Environmental scanning electron microscopy (ESEM)
EX30N peptide was dissolved in water to a final concentration of 1.5 mM. The solution was divided into vials that were sealed and kept at 50 °C into a thermostat for 1 h. 10 µl of the incubated solution was deposited on a silicon (100) wafer. Environmental scanning electron micrographs were recorded with a Philips Microscope (FEG source). Settings: ESEM dry mode with SE detector, PLA of 500 µm at a pressure of 1–3.5 Torr and at room temperature.Atomic force microscopy (AFM)
AFM experiments were carried out with a NanoScope III Multimode microscope (DI, California, USA) and with ultrasharp cantilevers (NT-MDT, Russia). Samples were dissolved in deionized water to a final concentration of 1.0 mg ml−1, and deposited as a droplet on a Si (100) wafer. After complete evaporation of the solvent, samples were attached to a metallic disc with double sided tape and analyzed in tapping mode.Results and discussion
CD spectroscopy
CD spectra of elastin peptides were recorded in two different solvents, water and trifluoroethanol (TFE).The CD spectrum of EX30N recorded in water at 0 °C shows typical features of PPII, a positive band at ca. 217 nm and a large negative band at 197 nm (Fig. 1a). Although many textbooks still state that the positive (217 nm)–negative (197 nm), non-conservative CD couplet is diagnostic of the random coil, an enormous amount of theoretical and experimental findings argue in favour of the PPII structure.26 Parenthetically, those recent demonstrations have finally done justice to the early suggestions of Tiffany and Krimm.27 Increasing the temperature reduces the contribution of the PPII conformation to the CD spectrum, as evidenced by a progressive disappearance of the band at 217 nm, and a related reduction as well as red shift of the band at 197 nm. Furthermore, the presence of an isoelliptic point at 203 nm clearly indicates a two state equilibrium. CD difference spectra were performed in order to highlight the nature of this transition, revealing the presence of a type I β turn favoured by higher temperature. CD spectra in TFE recorded at different temperatures show a common spectral profile, two negative bands at 202 nm and 220 nm (Fig. 1b). These data suggest the presence of predominant unorder together with β-turn(s).
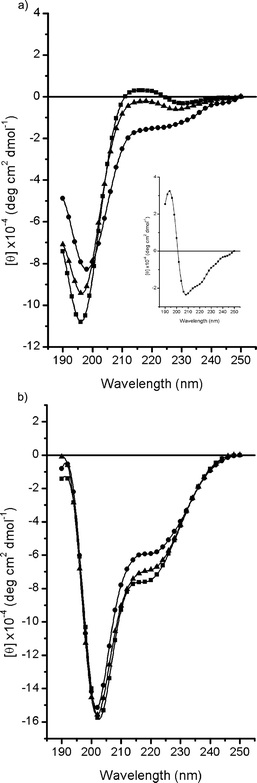 | ||
| Fig. 1 CD spectra of EX30N in (a) water and (b) TFE at different temperatures: 0 °C (■), 25 °C (▲) and 60 °C (●). The difference spectrum (60–0 °C) is also shown (inset in a). | ||
NMR spectroscopy
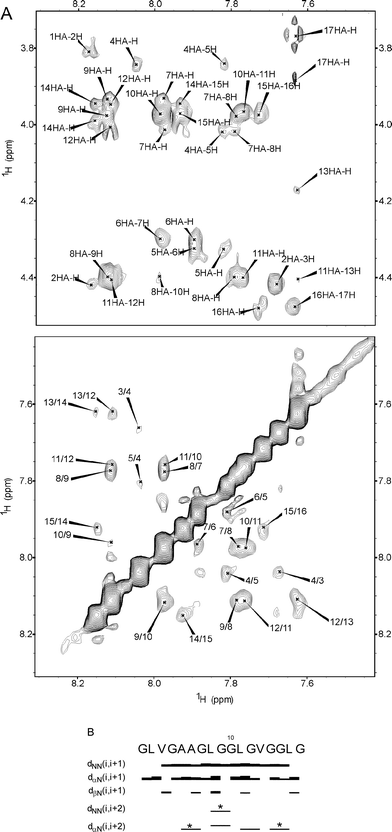
The EX30Npeptide was analyzed by 1H NMR spectroscopy in H2O–D2O (90 : 10) and in TFE-d3–H2O (80 : 20).Even though a high number of glycine residues (9 out of 17 residues) are present, a complete resonance assignment was achieved for all the peptide protons (Table SI and SII, ESI†). The NMR data of the sample recorded in H2O–D2O (90 : 10, v/v) exclude the presence of any folded structure. As a matter of fact, the chemical shifts of the Hα proton resonances are all in the range usually assigned to unordered conformations.28 No significant downfield shift is revealed, excluding the presence of a relevant amount of β-strand at 25 °C. The 3JNH-Hα coupling constants of non-glycine residues are in the range 5.8–7.6 Hz, usually assigned to unordered conformations, even though they are compatible also with PPII conformations. Only the valine residues show higher values, approximately 7.9Hz, suggesting a more extended conformation for these residues. While suggestive of the PPII conformation, as shown above by CD spectra, these NMR data alone are not able to define the presence of PPII. For this to occur, it is necessary to perform a careful NOE volume ratio analysis, between sequential dαN and dNN cross-peak volumes. As found by Fiebig et al.29 by statistical analysis, the random coil conformation should have an NOE ratio [αN(i,i + 1)/NN(i,i + 1] of 1.4 for the sequential backbone cross-peaks, while for the extended conformations the higher distance of sequential amide protons increases the ratio to a value of 55.30Fig. 2a shows the fingerprint (Hα-HN) and the amide (NH-NH) region of the ROESY spectrum of EX30N recorded in H2O–D2O (90 : 10) at 25 °C. The regions are displayed with the same threshold in order to highlight the difference in intensity of the cross-peaks. From the analysis of NOE volume, we were able to define the NOE ratio for some of not overlapping peaks (Table SIII, ESI†). The tabulated values show ratio values ranging from 0.97–11. In particular for the central region of the peptide, increased values are recorded (G7 = 11; L8 = 3.2; L11 = 3.6), suggesting that the time spent in the PPII conformation by the single residues at 25 °C is significant.
 | ||
| Fig. 2 The fingerprint region and the amide region of the ROESY spectra of EX30Npeptide recorded in H2O–D2O (90 : 10) at 298 K. The regions were displayed with the same threshold, in order to highlight the difference in intensity of the cross-peaks (A). Summary of sequential and medium range NOEs. The NOE intensities are reflected by the thickness of the line. * refers to overlapped peaks (B). | ||
The analysis of the EX30Npeptide in TFE-d3–H2O (80 : 20) by NMR spectroscopy provides evidence for the presence of several β-turns, most probably rapidly interconverting. As a matter of fact, reduced temperature coefficients for some residues were found; these findings indicate a reduced exposition to the solvent, usually ascribed to hydrogen bonding.31 In detail, G7, G10, L11, V13, G15,and L16 show temperature coefficients in the range of 4.0–4.5 ppb K−1, values usually attributed to very weak H-bonds or to an equilibrium between turn and other conformations devoid of H-bonds. In the case of β-turns, the lowered temperature coefficient is due to the presence of a hydrogen bond between the amide proton of the fourth residue and the carbonyl group of the first residue of the turn. Further evidence of turn structures are some intense sequential dNN(i,i + 1) values and typical medium range NOE [dαN(i,i + 2)] identified for residue L8-G10 and L11-V13 (Fig. 3). It was not possible to identify the typical medium range NOE [dαN(i,i + 2)] for all suggested turn structures, because of high overlap in the glycine residue region. In other cases, where overlap was not present, we could only suggest the presence of rapidly interconverting (sliding) β-turns, whose time spent in β-turn is reduced and consequently not revealed by the slow NMR time scale. The overall picture highlights a dynamic structural ensemble and reveals a good agreement with the CD results.
 | ||
| Fig. 3 The fingerprint region and the amide region of the NOESY spectra of EX30Npeptide recorded in TFE-d3–H2O (80 : 20) 289 K. (A). Summary of the NMR sequential and medium range NOEs. The NOE intensities are reflected by the thickness of the line. * refers to overlapped peaks (B). | ||
The presence of sliding β-turns in the general elastin repeat sequence XGGZG (with X, Z = V, L) is not surprising as they were previously ascertained for (LGGVG)n,32 and many other elastin-related polypeptides.33
FT-IR spectroscopy
The FT-IR spectrum of the amide I region of EX30N fibers (Fig. 4a) shows a prominent band at 1627 cm−1 due to antiparallel β-sheets of the cross-β type, together with a band at 1693 cm−1, which is typical of the antiparallel β-sheet conformation.34 Two other bands at 1643 cm−1 and 1672 cm−1 could originate from unordered + water and PPII conformations, respectively. The deconvoluted amide II region of EX30N fibers shows a component at 1516 cm−1 considered representative of the presence of β-sheet conformation, even though the amide II region is less informative regarding the secondary structures.35 The amide A spectrum is shown in Fig. 4b. It shows one strong band at 3306 cm−1, due to H-bonded NH amide stretching, together with two minor bands at 3444 and 3546 cm−1, assigned to H-bonded and free water O–H stretching. In order to remove the effect of water both on the amide I and amide A region we left the KBr pellet for 2 h at 50 °C before the acquisition of the spectrum. The dehydration shifts one band in the amide I region (Fig. 4c) from 1643 cm−1 to 1654 cm−1 which is now clearly attributable to the random coil. In the amide A region we note the disappearance of any band related to the OH stretching modes, thus confirming the full dehydration of the sample (Fig. 4d). | ||
| Fig. 4 Decomposed FT-IR spectra of EX30N fibers in KBr pellets. (a) Amide I and II region. (b) Amide A region. After dehydration (c) amide I and II regions show only slight variations, while (d) amide A region shows the absence of water-related OH stretchings. | ||
Molecular model of elastin-derived amyloid
The molecular studies performed in water on EX30N pointed toward a flexible ensemble of conformations with a not negligible number of PPII structures. This is not surprising in view of the relevant presence of glycine residues favouring both unordered and PPII conformations.It should be noted that the studies in aqueous solution exclude the presence of β-strands or β-sheets in significant amounts. Only FT-IR spectra of the aggregated sample revealed the presence of antiparallel β-sheets and cross-β structures. This observation prompted us to suggest a model for aggregation involving the PPII as the prefibrillar conformation.
The dihedral angles characterizing the PPII structure (ϕ = − 78; ψ = +150) are in a region of the Ramachandran map very close to that typical of β-sheet structures, and, consequently, only small variations are necessary for the transition to take place.
Accordingly, an “induced fit” model could be proposed (Fig. 5). The highly flexible monomers, populating significantly PPII conformations, would be expected to readily interact with each other or with preformed β-sheet seeds, by adapting slightly their dihedral angles. PPII has been proposed as the prefibrillar conformation in other amyloid-forming proteins,14,15,36 reinforced by recent demonstrations of significant amounts of PPII in the Alzheimer amyloid peptides, Aβ(1–28)37 and Aβ(12–28).38
 | ||
| Fig. 5 Simplified model of aggregation of elastin peptideEX30N. (i) PPII monomer interacts with preformed antiparallel β-sheet seeds, and adopts the β-sheet structure. (ii) PPII monomer aggregates with PPII antiparallel helices interacting through H- bonds. | ||
Alternatively, on the basis of our results, an aggregation model directly implying PPII conformations could not be completely ruled out. An example of this kind of aggregation pattern has been previously reported for (APG)n39 and (PGG)n.40 In these models PPII helices aggregated to form zigzag sheets, with neighboring antiparallel chains joined by interchain hydrogen bonding.41 The absence of proline residues in the EX30Npeptide could argue against this structure, nevertheless recent work of Ohnishi et al. revealed the presence of polyglycine II (PGII) structures (with dihedral angles, ϕ = −80; ψ = 150, identical to PPII) in the aggregated state of glycine-rich peptides. According to their NMR and small-angle X-ray scattering data the monomeric state shows direct evidence of PGII elongated structures, retained in the solid state of the aggregated samples. PGII favours the intermolecular association through backbone hydrogen bonding, thus prompting aggregation.
Turbidimetry experiments
EX30N peptide shows a great propensity to aggregate in water in an irreversible way, as monitored by turbidimetry experiments (Fig. 6), suggesting the formation of amyloid-like structures. The time course of aggregation is very slow at room temperature, while higher temperatures (for example 50 °C) are able to trigger the aggregation. Stirring of the solution is very important for the time course of the aggregation, as previously noted for other amyloid-forming proteins.42 Probably the alignment of the molecules, due to the fluid flow, renders their interaction in β-sheets more favourable, inducing the formation of seeds.14 | ||
| Fig. 6 Turbidimetry experiment on the EX30Npeptide, 4.0 mM in water. The turbidimetry on apparent absorbance (TAA) was recorded at λ = 440 nm as a function of time at room temperature (■) and at 50 °C (▲). | ||
Birefringence microscopy
One of the simplest way to ascertain the amyloid nature of peptide aggregate is Congo red staining.25 The property of amyloids to possess enhanced birefringence after staining with Congo red has been assigned to an ordered arrangement of the elongated stain molecules into the fibers, indicating that the aggregates are not amorphous but have a highly organized sub-structure.43 The observation of apple-green birefringence of the Congo red stained sample of EX30N analyzed under polarized light (Fig. 7) provides evidence for the amyloid nature of the aggregates. The apple-green birifrengence is evident at the border of the stained aggregate where less material is accumulated. | ||
| Fig. 7 Birefringence of Congo red stained EX30Npeptide fibers viewed by cross-polarization microscopy. (a) Bright field. (b) Cross-polarized light. | ||
Transmission electron microscopy (TEM)
Amyloid fibrils, in electron microscopy, typically appear as unbranched filaments that are a few nanometres in diameter and up to a micrometre or more in length. Sometimes, not always, they appear as helical, paired filaments. In order to confirm the amyloid nature of the sample, the EX30Npeptide was analyzed at a nanometre scale by TEM (transmission electron microscopy). The specimen was prepared at room temperature and immediately analyzed. Under these conditions the ultra-structural analysis by TEM revealed different aggregation profiles, going from dominantly amorphous aggregates to more complex fibrillar structures as shown in Fig. 8. The diameter of the fibrils are in the range commonly observed for amyloid fibers (6–12 nm). These structures show some flexibility, (Fig. 8a and c) and probably originated from the very short pseudo-filamentous sub-structures shown at higher magnification in Fig. 8b. The considerable bending and irregular forms of the short fibrils could be assigned to intermediates of the amyloid fibers where the stabilizing intermolecular interactions are still relatively weak as compared to the longer and more mature fibrils44 shown in Fig. 8d. These fibrils have a ribbon aspect and in some points are slightly twisted. Being that the irreversible aggregation is triggered by temperature, we analyzed the same peptide after 1 h incubation in aqueous solvent at 50 °C. A completely different macroscopic behavior during specimen preparation was observed, suggesting an evolution in the aggregation process during incubation. The formed fibers show a high tendency to stick together, forming viscoelastic globules. As a matter of fact, when a droplet of the peptide solution was withdrawn from the incubation solution, a viscoelastic consistency of the droplet with a high surface tension was evident by light microscopy. The sample was not observable by TEM, because of excessive staining. | ||
| Fig. 8 Room temperature self-assembly of EX30Npeptides in aqueous solution as amyloid-like filaments examined by TEM. (a) Amorphous aggregates with long fibrils; (b) short flexible protofilaments; (c) long fibrils; (d) after 2 months of incubation mature fibers with ribbon-like aspect are observed; samples were negatively stained by 1% uranyl acetate solution. | ||
In order to analyze the sample, we sonicated for 30 s the 1 h incubated solution prior to sample preparation. Sonication was able to break some of the unspecific bonds (weak surface interactions), thus allowing detailed analysis of the aggregates (Fig. 9). A fine network structure is revealed, composed of long unbranched rods, in some regions aligned in a parallel manner, in others irregularly spreading. While the diameter of the fibers did not change during incubation, an increase in intermolecular and inter-fiber interactions of a hydrophobic nature is suggested. As a matter of fact, the sequence of EX30N is formed exclusively by glycines and hydrophobic residues (A, L, V), prompting us to suggest that the only energetically favourable side-chain interactions able to stabilize the basic β-conformation of the amyloid fiber should be of hydrophobic nature. Increasing the temperature, analogously to coacervation of elastin,16,45 could induce the expulsion of water and trigger self-assemblyvia hydrophobic collapse.
 | ||
| Fig. 9 TEM image of EX30N sample sonicated after 1 h of incubation at 50 °C. A film of irregularly spread fibrils is revealed. The sample was negatively stained by 1% uranyl acetate solution. | ||
Environmental scanning electron microscopy (ESEM)
The TEM images of the EX30Npeptide, acquired at room temperature and after 1 h incubation at 50 °C revealed the starting and the ending point of a rather complex temperature-induced process, able to change the aggregation pattern of the EX30Npeptide. To characterize the intermediate stages of the aggregation, we examined the three dimensional characteristics of the evolving nanostructures by ESEM, where extensive dehydration does not occur.ESEM offers the opportunity to work on biological samples without complex and artifact-generating manipulations. Nevertheless, ESEM has been assigned a reduced importance in amyloid ultra-structural analysis, mainly carried out by TEM and AFM (atomic force microscopy). Only a few studies on amyloid-forming proteins and peptides have conducted by ESEM, showing reticular,46 fibrillar,47 and spherical48 higher-order supramolecular structures.
The sample was analyzed by ESEM immediately after withdrawal from the solution. The initial images of EX30N show the presence of globules of uniform size (diameter, approximately 0.5–0.7 µm) (Fig. 10). These almost spherical particles are closely packed and evolve after 20 min incubation at 50 °C to highly flexible protofibrils, prone to forming a thin film. After 40 min of incubation a complex reticular network is formed. The connections between the fibrils increase and a developing film covering the network was observable in several sites.
 | ||
| Fig. 10 ESEM micrographs of EXN30peptide incubated at 50 °C at different incubation times. (a) Time = 0, closely packed globules of ca. 0.5–0.7 µm diameter; (b) t = 20 min, flexible protofibrils; (c) t = 40 min, a complex reticular network; (d) t = 70 min, a dense viscoelastic film with starting globules in the background. (e) t = 19 h and (f) t = 6 d, a compact film. | ||
After 70 min, in many points of the specimen a dense viscoelastic film has formed. In the background the starting globules are still evident. After 19 h the formation of a compact film is witnessed by the presence of large bubbles coming out from the peptide film. In some points holes are visible where the water bubbles escaped from the surface.
Atomic force microscopy (AFM)
The presence of globules as starting material is evident also in the AFM micrographs (Fig. 11). In this case some protofibrillar structures are also evident, coming out from the aligned juxtaposition of the globules, a behavior similar to that of another elastin sequence, able to form amyloids, that is the exon28-coded domain of elastin.47 Also in that case globules are the precursor for intertwined filaments of defined diameter and pitch. Nevertheless, in the case of EX30Npeptide, the amyloid-like fibers observed in Fig. 11a evolve further after incubation at 50 °C into a film incorporating globules as observed in ESEM. | ||
| Fig. 11 AFM images of EX30Npeptide. (a) At room temperature, elongated structures are present; b) after 1 h incubation at 50 °C. | ||
Conclusions
In this paper we report on the molecular and supramolecular characterization of an amyloid-forming elastin peptide.The molecular studies highlight the important role of PPII conformations for the prefibrillar state, suggesting that aggregation takes place through addition of the monomer in the PPII conformation with the preformed β-sheet aggregate.
Some authors proposed glycine residues as an evolutionary strategy to avoid amyloid aggregation,49 our data conversely suggest that the presence of a high number of glycine residues, prompting the adoption of PPII conformation, can induce amyloid fiber formation either through cross-β structures or directly through interaction of PPII helices. Obviously, the two processes are not necessary mutually exclusive. Once again the simplest amino acid in nature, glycine, shows a highly flexible behavior that could not be easily rationalized.
The supramolecular studies revealed a complex nano-aggregation profile that starting from globules evolve further in a compact film through an amyloid aggregation mechanism. For the first time this evolution has been monitored in detail by different microscopy techniques, that is TEM, AFM, and mainly ESEM, pointing out a complex self-assembling mechanism where globules are the starting point for the production of a thin nano-film. Some other amyloid-forming proteins are able to form spherical structures, called spherulites or simply spherical aggregates formed after proper evolution, often as an alternative aggregation pathway.20 Conversely, EX30N elastin peptide shows spherical aggregates, composed of amyloid fibrils, as a starting point, evolving later into a complex three-dimensional network, which cumulates in a film layer, obtained by side-by-side interacting fibrils.
These peptide-nanostructures have many potential applications in various fields including tissue engineering, and materials science. For instance, the propensity to form a thin film could be exploited for a biocompatible coating of artificial grafts, in order to improve the performance and potency of small-diameter vascular grafts. Further studies in this direction are still in progress.
Acknowledgements
We thank Dr Angelo De Stradis for TEM micrographs. Mr Alessandro Laurita for TEM and ESEM micrographs. Dr Roberta Flamia for AFM images. The financial support of EU (Elastage - Contract no. 018960) is gratefully acknowledged.References
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS; F. J. Arias, V. Reboto, S. Martin, I. Lopez and J. C. Rodriguez-Cabello, Biotechnol. Lett., 2006, 28, 687–695 CrossRef CAS.
- J. E. Wagenseil and R. P. Mecham, Birth Defects Res., Part C: Embryo Today, 2007, 81, 229–240 Search PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS.
- D. M. Fowler, A. V. Koulov, W. E. Balch and J. W. Kelly, Trends Biochem. Sci., 2007, 32, 217–224 CrossRef CAS.
- I. Cherny and E. Gazit, Angew. Chem., Int. Ed., 2008 Search PubMed; H. Yan, A. Saiani, J. E. Gough and A. F. Miller, Biomacromolecules, 2006, 7, 2776–2782 CrossRef CAS.
- A. Pepe, B. Bochicchio and A. M. Tamburro, Nanomedicine (London, U. K.), 2007, 2, 203–218 Search PubMed.
- A. W. Clarke, E. C. Arnspang, S. M. Mithieux, E. Korkmaz, F. Braet and A. S. Weiss, Biochemistry, 2006, 45, 9989–9996 CrossRef CAS.
- G. M. Bressan, P. Argos and K. K. Stanley, Biochemistry, 1987, 26, 1497–1503 CrossRef CAS.
- K. Trabbic-Carlson, L. A. Setton and A. Chilkoti, Biomacromolecules, 2003, 4, 572–580 CrossRef CAS; K. Nagapudi, W. T. Brinkman, B. S. Thomas, J. O. Park, M. Srinivasarao, E. Wright, V. P. Conticello and E. L. Chaikof, Biomaterials, 2005, 26, 4695–4706 CrossRef CAS.
- C. M. Bellingham, M. A. Lillie, J. M. Gosline, G. M. Wright, B. C. Starcher, A. J. Bailey, K. A. Woodhouse and F. W. Keeley, Biopolymers, 2003, 70, 445–455 CrossRef CAS.
- R. Flamia, P. A. Zhdan, M. Martino, J. E. Castle and A. M. Tamburro, Biomacromolecules, 2004, 5, 1511–1518 CrossRef CAS; L. L. del Mercato, G. Maruccio, P. P. Pompa, B. Bochicchio, A. M. Tamburro, R. Cingolani and R. Rinaldi, Biomacromolecules, 2008, 9, 796–803 CrossRef CAS; B. A. Kozel, H. Wachi, E. C. Davis and R. P. Mecham, J. Biol. Chem., 2003, 278, 18491–18498 CrossRef CAS.
- J. C. Rodriguez-Cabello, Adv. Exp. Med. Biol., 2004, 553, 45–57 CAS; K. Channon and C. E. MacPhee, Soft Matter, 2008, 4, 647–652 RSC; A. Chilkoti, T. Christensen and J. A. MacKay, Curr. Opin. Chem. Biol., 2006, 10, 652 CrossRef CAS.
- M. Calamai, F. Chiti and C. M. Dobson, Biophys. J., 2005, 89, 4201–4210 CrossRef CAS.
- C. D. Syme, E. W. Blanch, C. Holt, R. Jakes, M. Goedert, L. Hecht and L. D. Barron, Eur. J. Biochem., 2002, 269, 148–156 CrossRef CAS.
- E. W. Blanch, L. A. Morozova-Roche, D. A. Cochran, A. J. Doig, L. Hecht and L. D. Barron, J. Mol. Biol., 2000, 301, 553–563 CrossRef CAS.
- A. Pepe, D. Guerra, B. Bochicchio, D. Quaglino, D. Gheduzzi, I. Pasquali Ronchetti and A. M. Tamburro, Matrix Biol., 2005, 24, 96–109 CrossRef CAS.
- A. M. Tamburro, A. Pepe, B. Bochicchio, D. Quaglino and I. P. Ronchetti, J. Biol. Chem., 2005, 280, 2682–2690 CAS.
- B. Bochicchio, A. Pepe and A. M. Tamburro, Chirality, 2008 Search PubMed.
- D. G. Davis and A. Bax, J. Am. Chem. Soc., 1985, 107, 2820–2821 CrossRef CAS.
- J. Jeener, B. H. Meier, P. Bachmann and R. R. Ernst, J. Chem. Phys., 1979, 71, 4546–4553 CrossRef CAS.
- T. L. Hwang and A. J. Shaka, J. Magn. Reson., Ser. A, 1995, 112, 275–279 CrossRef CAS.
- F. Delaglio, S. Grzesiek, G. W. Vuister, G. Zhu, J. Pfeifer and A. Bax, J. Biomol. NMR, 1995, 6, 277–293 CrossRef CAS.
- T. D. Goddard and D. G. Kneller, Sparky, University of California, San Francisco Search PubMed.
- K. Wüthrich, NMR of Proteins and Nucleic Acids, Wiley, New York, 1986 Search PubMed.
- M. R. Nilsson, Methods, 2004, 34, 151–160 CrossRef CAS.
- B. Bochicchio and A. M. Tamburro, Chirality, 2002, 14, 782–792 CrossRef CAS; R. W. Woody, Adv. Biophys. Chem, 1992, 2, 37–79 Search PubMed; Z. Shi, R. W. Woody and N. R. Kallenbach, Adv. Protein Chem., 2002, 62, 163–240 CAS.
- M. L. Tiffany and S. Krimm, Biopolymers, 1968, 6, 1379–1382 CAS.
- D. S. Wishart, B. D. Sykes and F. M. Richards, Biochemistry, 1992, 31, 1647–1651 CrossRef CAS.
- K. M. Fiebig, H. Schwalbe, M. Buck, L. J. Smith and C. M. Dobson, J. Phys. Chem., 1996, 100, 2661–2666 CrossRef CAS.
- G. J. Sharman and M. S. Searle, J. Am. Chem. Soc., 1998, 120, 5291–5300 CrossRef CAS.
- G. D. Rose, L. M. Gierasch and J. A. Smith, in Advanced Protein Chemistry, ed. C. B. Anfinsen, J. T. Edsall and F. M. Richards, Academic Press, New York, 1985, vol. 37, p. 1–109 Search PubMed.
- A. M. Tamburro, in Elastin: Chemical and Biological Aspects, ed. A. M. Tamburro and J. M. Davidson, Galatina Congedo Editore, Potenza, 1990, pp. 127–145. Search PubMed; A. M. Tamburro, V. Guantieri and D. D. Gordini, J. Biomol. Struct. Dyn., 1992, 10, 441–454 Search PubMed.
- A. M. Tamburro, B. Bochicchio and A. Pepe, Biochemistry, 2003, 42, 13347–13362 CrossRef CAS.
- M. Jackson and H. H. Mantsch, Crit. Rev. Biochem. Mol. Biol., 1995, 30, 95–120 CrossRef CAS; W. H. Moore and S. Krimm, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 4933–4935 CAS.
- J. Bandekar, Biochim. Biophys. Acta, 1992, 1120, 123–143 CAS.
- E. W. Blanch, A. C. Gill, A. G. Rhie, J. Hope, L. Hecht, K. Nielsen and L. D. Barron, J. Mol. Biol., 2004, 343, 467–476 CrossRef CAS.
- F. Eker, K. Griebenow and R. Schweitzer-Stenner, Biochemistry, 2004, 43, 6893–6898 CrossRef CAS.
- J. Jarvet, P. Damberg, J. Danielsson, I. Johansson, L. E. Eriksson and A. Graslund, FEBS Lett., 2003, 555, 371–374 CrossRef CAS.
- B. B. Doyle, W. Traub, G. P. Lorenzi and E. R. Blout, Biochemistry, 1971, 10, 3052–3060 CrossRef CAS.
- W. Traub, J. Mol. Biol., 1969, 43, 479–485 CrossRef CAS.
- D. M. Segal and W. Traub, J. Mol. Biol., 1969, 43, 487–496 CrossRef CAS.
- L. C. Serpell, J. Berriman, R. Jakes, M. Goedert and R. A. Crowther, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 4897–4902 CrossRef CAS; T. R. Serio, A. G. Cashikar, A. S. Kowal, G. J. Sawicki, J. J. Moslehi, L. Serpell, M. F. Arnsdorf and S. L. Lindquist, Science, 2000, 289, 1317–1321 CrossRef CAS; E. K. Hill, B. Krebs, D. G. Goodall, G. J. Howlett and D. E. Dunstan, Biomacromolecules, 2006, 7, 10–13 CrossRef CAS.
- V. N. Uversky, Nanomedicine (London, U. K.), 2007, 2, 615–643 Search PubMed.
- T. P. Knowles, A. W. Fitzpatrick, S. Meehan, H. R. Mott, M. Vendruscolo, C. M. Dobson and M. E. Welland, Science, 2007, 318, 1900–1903 CrossRef CAS.
- C. M. Bellingham, K. A. Woodhouse, P. Robson, S. J. Rothstein and F. W. Keeley, Biochim. Biophys. Acta, 2001, 1550, 6–19 CAS.
- M. J. Krysmann, V. Castelletto and I. W. Hamley, Soft Matter, 2007, 3, 1401–1406 RSC.
- B. Bochicchio, A. Pepe, R. Flamia, M. Lorusso and A. M. Tamburro, Biomacromolecules, 2007, 8, 3478–3486 CrossRef CAS.
- M. R. H. Krebs, G. L. Devlin and A. M. Donald, Biophys. J., 2007, 92, 1336–1342 CAS; M. R. Krebs, C. E. Macphee, A. F. Miller, I. E. Dunlop, C. M. Dobson and A. M. Donald, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14420–14424 CrossRef CAS.
- E. Monsellier and F. Chiti, EMBO Rep., 2007, 8, 737–742 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Proton resonance assignments of EX30N in H2O–D2O (90 : 10) and in TFE-d3–H2O (80 : 20); NOE ratio analysis. See DOI: 10.1039/b811286j |
| This journal is © The Royal Society of Chemistry 2009 |