Independent tuning of multiple biomaterial properties using protein engineering†
Karin S.
Straley
a and
Sarah C.
Heilshorn
*b
aChemical Engineering Department, Stanford University, 466 Lomita Mall, Moore Building, Room 271, 94305-4045, USA
bMaterials Science and Engineering Department, Stanford University, 476 Lomita Mall, McCullough Building, Room 246, Stanford, CA 94305-4045, USA. E-mail: heilshorn@stanford.edu; Fax: +1-650-498-5596; Tel: +1-650-723-3763
First published on 23rd October 2008
Abstract
A key attribute missing from many current biomaterials is the ability to independently tune multiple biomaterial properties without simultaneously affecting other material parameters. Because cells are well known to respond to changes in the initial elastic modulus, degradation rate, and cell adhesivity of a biomaterial, it is critical to develop synthetic design strategies that allow decoupled tailoring of each individual parameter in order to systematically optimize cell-scaffold interactions. We present the development of a biomimetic scaffold composed of chemically crosslinked, elastin-like proteins designed to support neural regeneration through a combination of cell adhesion and cell-induced degradation and remodeling. The design of these engineered proteins includes cell adhesion sequences to enable neuronal attachment as well as sequences sensitive to cleavage by urokinase plasminogen activator (uPA), a protease locally secreted from the tips of growing neurites, to enable highly localized and tunable degradation properties. These engineered proteins are produced using recombinant techniques and chemically crosslinked into highly swollen hydrogels with controllable mechanical properties. Through a modest 3% change in the chemical identity of three otherwise identical engineered proteins, we can modify the uPA substrate specificity resulting in tunable changes in protease degradation half-life over two orders of magnitude. Under high uPA exposure, the designed scaffolds exhibit systematic variation of scaffold lifetime, from being fully degraded within a single day to showing no noticeable degradation within a full week. In vitro studies using the model PC-12 neuronal-like cell line show that the crosslinked proteins support tunable cell adhesion and neuronal differentiation. Increasing the density of RGD peptides present in the protein substrates leads to increased cell adhesion and more extensive neurite outgrowth. These engineered proteins offer the ability to independently tailor the mechanics, degradation properties, and cell adhesivity of scaffolds for the study of central nervous system regeneration.
Introduction
The treatment of central nerve injuries presents a particularly difficult challenge in medicine today. Every year in the United States, around 11,000 spinal cord injuries occur.1,2 At present, there are no clinical repair strategies that promote significant functional recovery following traumatic central nervous system (CNS) injury. Upon injury, the native cells and proteins in the CNS form a complex, inhibitory scar that essentially blocks nerve growth while the injured cells release biochemicals that propagate further nerve damage.3 Despite the inherent challenges presented by central nerves, they have been observed to partially regenerate through peripheral nerve grafts.4 A combined therapy involving implantation of a biomaterial scaffold supplemented with cells and growth factors to mimic the restorative capacity of the peripheral nervous system is hypothesized to be the most promising approach to CNS treatment.3,5Biodegradable materials are the preferred choice for construction of CNS regeneration scaffolds.3,6–8 These materials eliminate complications resulting from a permanent implant, such as compression of regenerating nerves or a prolonged inflammatory response, which may necessitate a second surgery for removal.3,7,9 In addition, biodegradable materials aim to promote nerve growth by providing a less obstructive regeneration environment that can be replaced by cells and their matrix.7,8 The most common biodegradation strategy involves materials that degrade uniformly over time at the bulk level due to hydrolysis.6,10–12 For example, by creating PEG-PLA scaffolds with faster degradation kinetics, the outgrowth of neural processes was shown to be enhanced.13 In contrast, natural scaffolds such as chitosan, fibrin, and collagen degrade in response to enzymes prevalent in the extracellular matrix (ECM).14–16 Similar to the synthetic scaffolds, enhanced fibrin matrix degradation via enzymatic action of serine proteases has been linked to increased neurite extension.14 Taken together, these results suggest that tailoring the rate of biomaterial degradation may be a useful strategy to control the rate of neuronal outgrowth in CNS regeneration scaffolds.
Synthetic mimics of the naturally degradable ECM environment have been designed by incorporating short, protease-sensitive peptide sequences into synthetic polymeric scaffolds.17–20 By increasing the protease sensitivity of these polymers to matrix metalloproteinase, the outgrowth of fibroblast cells and migration of osteoblasts have been enhanced.19–21 A key advantage of this technique over traditional hydrolytic degradation is that the biomaterial can respond to local changes in cellular behavior, which may vary from patient to patient and site to site, while hydrolytic cleavage cannot be tuned post-implantation. However, because the protease-sensitive peptide sequences are also used as crosslinking sites within these scaffolds, the degradation properties and the initial mechanical properties of these materials cannot be independently tuned.
Because neural cells are known to respond to the mechanical properties,22–25 degradation properties,13,14 and cell adhesive properties of scaffolds,26–29 our goal is to design a family of biomaterials that allows independent tuning of all three variables. Current biomaterials, including synthetic polymeric matrices, harvested natural biopolymers, and synthetic/natural hybrids, generally cannot independently tune the initial mechanical properties, degradation rate, or cell adhesivity without simultaneously affecting other material properties.30,31 For CNS nerve grafts, it is particularly important to fabricate a scaffold that allows neurons to locally degrade a material at the site of regeneration while simultaneously maintaining the bulk structural integrity of the implant. This approach is aimed at enhancing neurite outgrowth and preventing material collapse, which has been a confounding factor in many biodegradable peripheral nervous system and CNS guidance channels.32–34
To achieve this goal, we have designed a scaffold composed entirely of engineered proteins that mimics many of the essential characteristics of the natural ECM (Fig. 1). To our knowledge this is the first application of full length engineered proteins in the construction of nerve regeneration scaffolds. Recent work with self-assembled peptide amphiphiles has demonstrated the efficacy of using peptide containing materials to inhibit glial scar formation and promote neurite outgrowth in mouse spinal cords.26,27 Our protein polymers are synthesized using recombinant protein technology in order to exercise exact molecular-level control over the polymer composition. In addition, protein materials release no cytotoxic degradation fragments, can easily incorporate bioactive functionalities, and can be chemically crosslinked to form hydrogel scaffolds.35–37 A key advantage of recombinant protein engineering is the modular design and templated synthesis that allows creation of an entire family of polymers with similar amino acid sequences and precisely altered material properties. Here we demonstrate this advantage by designing a family of biomaterials with independently tunable mechanical properties, degradation rates, and cell adhesivity.
 | ||
| Fig. 1 Amino acid sequences of the engineered proteins (EK refers to enterokinase, ↑ refers to the predicted uPA protease cut site). | ||
Specifically, the engineered protein polymers detailed in this paper are composed of alternating elastin-like structural sequences and bioactive peptide sequences.38–40 To promote cell adhesion, the extended RGD sequence of fibronectin was designed into the protein. This sequence has been previously shown to promote neurite attachment due to interaction with integrins displayed on the surfaces of neurites.41 To induce local neuronal remodeling of the engineered protein, protease cleavage sequences of varying sensitivity to the enzyme urokinase plasminogen activator (uPA) were designed into the protein. The uPA enzyme is a member of the serine protease family, has been shown to be locally secreted from the tips of neuronal growth cones, and is thought to provide a mechanism for remodeling the natural ECM.42,43 We hypothesize that neurons may be able to degrade these engineered proteins specifically at the site of neurite elongation to create tunnels for neurite extension without compromising the bulk integrity of the scaffold. By varying the rate of scaffold degradation, it may be possible to influence the speed of neurite elongation to promote nerve regeneration.
These materials are intended for eventual use as pre-formed, implantable nerve grafts, either with or without the pre-seeding of exogenous cells. This type of implant is designed to link severed nerves to their original contacts, serving as a bridge across the inhibitory CNS injury site. In the peripheral nervous system, nerve grafts are commonly used to provide a regenerative growth environment and guidance for growing neurons.44–46 Initial successes using peripheral nerve graft implants in the CNS have led to substantial interest in designing artificial, cell-free nerve grafts for the inhibitory CNS environment.4,45
In this paper, our engineered protein polymers are shown to be easily produced in Escherichia coli bacteria, purified, and chemically crosslinked into protein scaffolds with tunable elastic moduli. The protease sensitive proteins are shown to exhibit controlled degradation in both their uncrosslinked and crosslinked forms. A 3% change in monomer sequence at the protease cleavage site was shown to result in a greater than 200-fold change in protease degradation half-life of the uncrosslinked protein polymers. This same amino acid change was also shown to result in crosslinked films that ranged from fully degraded at one day to virtually non-degradable after seven days of constant uPA exposure. In vitro studies using the model PC-12 neuronal-like cell line indicate that crosslinked protein scaffolds containing the RGD domain from fibronectin support cell adhesion, neuronal differentiation, and neurite extension. We demonstrate direct control over the cell adhesivity and neurite outgrowth by tuning the density of RGD peptides within the scaffold.
Unlike most biomaterials, this new family of scaffolds has the distinct advantage of allowing the initial mechanical properties, the degradation properties, and the cell adhesive properties to be tailored independently without adversely affecting the other material properties. These biomaterials provide an ideal framework for conducting systematic studies into the optimal scaffold material properties required for CNS repair.
Experimental
Plasmid construction
A schematic of the plasmid construction process is shown in the supporting information. Oligonucleotides that encode the three protein domain sequences (tag, bioactive, and structural elastin-like domains) were purchased from Integrated DNA Technologies (Coralville, IA). The coding and anticoding oligonucleotide strands for each domain were phosphorylated and annealed to create the following DNA overhang sequences on the 5′ and 3′ ends: tag domain (NcoI and XhoI), bioactive domains (EcoRI and HindIII), and elastin-like structural domain (KpnI and KpnI).The tag domain was ligated into pET15b (Novagen, San Diego, CA) to form Tag-pET15b. The bioactive domains were ligated into pUC18 to form Bio-pUC18. The elastin-like monomer domain was multimerized using T4 DNA ligase at 4 °C for 3 min and the trimer reaction product was isolated by gel electrophoresis and ligated into Bio-pUC18 to form Bio-[Elastin]3-pUC18. The bioactive and elastin-like domains were removed from Bio-[Elastin]3-pUC18 with SalI and XhoI and ligated into Tag-pET15b digested with XhoI to form Tag-[Bio-[Elastin]3]-pET15b. This reaction was repeated three times to form Tag-[Bio-[Elastin]3]4-pET15b, and the final plasmid content was confirmed by sequencing (Stanford PAN Facility).
Protein expression and purification
Individual Tag-[Bio-[Elastin]3]4-pET15b plasmids were transformed into the Escherichia coli expression strain BL21(DE3). Protein expression was induced under the control of the T7-lac promoter at an OD600 of 0.6 (37 °C, LB medium) with 1 mM β-isopropyl thiogalactoside and allowed to express for 3 hrs post induction. The wet cell pellet was resuspended in TEN Buffer (1 g/mL, 1 mM PMSF), sonicated, and agitated overnight at 4 °C. The pH was adjusted to ∼9 with 4 N NaOH, incubated at 4 °C for 1 h, and centrifuged at 4 °C (1 h, 17,700 g). NaCl was added to the supernatant to a final concentration of 1 M. This solution was agitated overnight at 4 °C, incubated at 37 °C shaking for 3 hrs, and centrifuged at 37 °C (1 h, 3,584 g). The pellet was resuspended in water (0.1 g/mL), agitated overnight at 4 °C, adjusted to pH ∼9 with 1 N NaOH, incubated at 4 °C shaking for 1 h, and centrifuged at 4 °C (1 h, 34,929 g). Warm (37 °C, 1 M NaCl) and cold (4 °C, pH ∼9) purification cycles were each repeated twice. The final supernatant was desalted using size exclusion centrifugation (10,000 MWCO, Millipore Amicon, Billerica, MA) and lyophilized. Typical protein yields were 25–50 mg/L.Amino acid analysis
Amino acid analysis was performed by the Molecular Structure Facility at the University of California, Davis (Davis, CA) using a Hitachi L-8800 sodium citrate-based amino acid analyzer (Tokyo, Japan). Protein samples were cleaved by HCl hydrolysis, separated with ion-exchange chromatography, and detected using a ninhydrin reaction.Lower critical solution temperature
Lyophilized protein was resuspended at a concentration of 10 mg/mL in phosphate buffered saline (PBS, pH = 7.2, 4 °C). Absorbance readings at 300 nm were monitored as a function of temperature using a Molecular Devices SpectraMax Plus384 Spectrophotometer (Sunnyvale, CA). The temperature was increased at a rate of 0.1 °C/min with an equilibration time of 30 s at each step.Scaffold crosslinking and material characterization
Crosslinked hydrogels were made by dissolving lyophilized protein in PBS buffer (pH = 7.4) at a concentration of 0.1 mg/µL and rapidly mixing in disuccinimidyl suberate (DSS, Pierce Biotechnology, Rockford, IL) solubilized in 25:75 dimethyl formamide: dimethyl sulfoxide solution (0.042 mg DSS/µL of solvent) at 4 °C. The mixed solution was compressed between two glass plates covered with parafilm (separated with 1 mm spacers) and allowed to react at room temperature for 24 hrs (reaction time is much less than 24 hrs but allowing the samples to dry for 1 day minimized loss of sample integrity due to adherence to the mold). Samples were crosslinked at stoichiometric ratios of 0.75–1.5:1 NHS esters to primary amines. Crosslinked hydrogels were cut into 5 mm diameter circles (samples were 400–600 µm thick).The compressive elastic modulus of each protein was determined in triplicate using a TA AR2000 Rheometer (New Castle, DE), compressive rate of 2 µm/s, 22 °C, in PBS buffer (pH = 7.4). Compressive elastic moduli (E) were determined by measuring the slope of the initial linear portion of the stress-strain curve (0–15% strain).
The average molecular weight between crosslinks (Mc) was estimated using rubber elasticity theory according to Equation 1, where ρ is the protein density (approximated as 1.35 g/mL),47 R is the ideal gas constant, and T is temperature.48 Mc was used to estimate the percentage of lysines reacted during protein crosslinking.
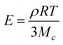 | (1) |
The water content (W) was measured in duplicate using Equation 2, where ms is the weight of the hydrated sample and md is the weight of the dehydrated sample. Dehydrated material was prepared using a vacuum oven operated at 50 °C for 24 hrs.
 | (2) |
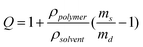
The volumetric swelling ratio (Q) was determined in duplicate using Equation 3, where ρpolymer is the density of the protein polymer (approximated as 1.35 g/mL) and ρsolvent is the density of the solvent (approximated as 1.0 g/mL).
 | (3) |
Protease degradation of uncrosslinked proteins
The degradation reactions were carried out in triplicate at 37 °C (200 µL reaction volume, 0.1 µM (528 U/mL) with low molecular weight uPA (Calbiochem, San Diego, CA) and 100 µM engineered protein in sodium borate buffer (pH = 8)). Samples (6 µL) were taken at specific time points and immediately frozen on dry ice in a solution containing water (9 µL) and 3x SDS sample buffer (7.5 µL). The extent of protease degradation was monitored by separating each sample by gel electrophoresis (12% SDS-PAGE), staining the degradation fragments with Coomassie Brilliant Blue, and performing densitometry analysis (NIH ImageJ software, Bethesda, MA).Protein bands were transferred to a PVDF membrane in 1x CAPS buffer (100 mA, for 1 h, at 4 °C). The membranes were stained with Coomassie for 45 s and destained (45% methanol, 45% water, 10% acetic acid) for ∼1 h. The desired bands were cut from the membrane and submitted for N-terminal Edman degradation sequencing at the Stanford PAN Facility (Stanford, CA).
Kinetic analysis of protein degradation
Solutions of engineered proteins were made in sodium borate buffer (pH = 8, 4 °C) at the following concentrations: 0, 50, 100, 150, 200, 250, 300, and 350 µM. Solutions were prepared in triplicate with a total reaction volume of 260 µL. The protease degradation reactions were carried out at 37 °C with mixed high and low molecular weight uPA (Calbiochem, San Diego, CA, 0.27 U/µL). Samples (30 µL) were taken at specific time points for the u1 protein (0, 5, 10, 20, 30, 40, and 50 min) and the u2 protein (0, 0.5, 1, 2, 3, 4, and 5 h). These samples were pipetted immediately into a 96-well plate chilled on dry ice. In addition to samples from the degradation reactions, each 96-well plate contained calibration samples of u1 and u2 protein solutions of known concentrations (0, 50, 100, 150, 200, 250, 300, and 350 µM) in sodium borate buffer (pH = 8).All samples were thawed at 4 °C, and 10 µL of sodium borate buffer and 20 µL of 0.05% 2,4,6-trinitrobenzene sulfonic acid (TNBSA, Pierce Biotechnology, Rockford, IL) were added to each well. The TNBSA was allowed to react with the primary amines contained in each sample at 4 °C with agitation for ∼1 h. The absorbance at 425 nm was determined for all samples using a Molecular Devices SpectraMax Plus384 Spectrophotometer (Sunnyvale, CA). The kinetic data was fit to the Michaelis-Menten equation by applying an iterative Levenberg-Marquardt algorithim using KaleidaGraph version 4.03 software (Synergy Software, Reading, PA).
Protease degradation of protein films
Crosslinked hydrogels were made as described above with a 1:1 stoichiometric ratio of NHS esters to primary amines. Crosslinked hydrogels were cut into 5 mm diameter circles. The samples were degraded in PBS buffer (pH = 7.4) at room temperature under constant agitation with 300 U/mL of mixed high and low molecular weight uPA (Calbiochem, San Diego, CA). The compressive elastic modulus of each protein was determined in triplicate at the specified time points using a TA AR2000 Rheometer (New Castle, DE), compressive rate of 2 µm/s, 22 °C, in PBS buffer (pH = 7.4). Compressive elastic moduli were determined by measuring the slope of the initial linear portion of the stress-strain curve (0–15% strain).PC-12 cell culture conditions
PC-12 cells (ATCC, Manassas, VA) were grown in F12 Kaighn's complete media (10% horse serum, 5% fetal bovine serum, and 1% penicillin-streptomycin) at 37 °C, 5% CO2. Cells were differentiated in F12 Kaighn's differentiation media (1% penicillin-streptomycin, 50 ng/mL recombinant human β-NGF (R&D Systems, Minneapolis, MN)) at 37 °C, 5% CO2. Media was changed every two days for all cell experiments.Calcein AM cell adhesion assay
PC-12 cell adhesion was compared on glass coverslips coated with adsorbed collagen, plain coverslips, and coverslips spincoated with crosslinked protein films. Solutions containing a final protein concentration of 5 mg/mL were prepared at 4 °C by mixing 14 µL of protein dissolved in PBS (pH = 7.4) with 1 µL of 0.22 µg/µL DSS in 25:75 DMF:DMSO (1:1 reaction stoichiometry of NHS esters to amines). These solutions (15 µL) were pipetted onto 12 mm glass coverslips, spread across the surface, and spun at 5,200 rpm for 1.5 min at 4 °C using a Laurell Technologies Corporation Model WS-400B-6NPP/LITE spincoater. The films were allowed to crosslink, air dried at room temperature for 24 hrs, sterilized under UV light for 20 min, and soaked in PBS solution at 4 °C overnight to wash away unreacted protein and crosslinker. Crosslinked protein substrates included films of the RDG and RGD proteins as well as 1:1 mixtures of RGD:u1, RGD:u2, RGD:u3, and RGD:RDG.To prepare collagen control surfaces, glass coverslips (12 mm diameter) were sterilized with 100% ethanol for 20 min, rinsed three times with cold PBS, and incubated with rat tail collagen I (50 µg/mL in PBS, Sigma-Aldrich (St. Louis, MO)) overnight at 4 °C.
All coverslips (including plain glass controls) were then rinsed three times with PBS, blocked with BSA (fraction V, 0.2% in PBS, heat inactivated at 85 °C for 10 min) to prevent non-specific binding, and rinsed three more times with PBS.
PC-12 cells (passage 6) were plated onto films at a density of 10,000 cells/cm2 and cultured in F12 Kaighn's complete media for 6 days. Cultures were then washed 1x with PBS buffer, stained with 1.5 µM Calcien AM in PBS (Invitrogen, Carlsbad, CA), and fluorescently imaged (20 fields per sample, field size = 0.36 mm2, in triplicate) with an inverted Zeiss Axiovert 200 microscope (Oberkochen, Germany, 20x objective) and a CCD camera. Statistical significance between data sets was determined using a paired, two-tailed Student's t-test.
AlamarBlue® cell adhesion assay
Crosslinked protein films (1:1 u1:RGD, u1:RDG, u2:RGD, u2:RDG, u3:RGD, u3:RDG, RDG:RGD, and full RGD and RDG) were prepared on the surface of a 48-well polystyrene tissue culture plate. Crosslinking solutions containing a final protein concentration of 5 mg/mL were prepared at 4 °C by mixing 93.3 µL of protein dissolved in PBS (pH = 7.4) with 6.7 µL of 0.22 µg/µL DSS in 25:75 DMF:DMSO (1:1 reaction stoichiometry of NHS esters to amines). These solutions (100 µL) were pipetted onto the surface of each well. The 48-well plate was centrifuged at 3,000 g to ensure coverage of entire well surface. Excess solution was removed and films were then reacted and dried at room temperature for 24 hrs, sterilized under UV light for 20 min, and soaked in PBS solution at 4 °C overnight to wash away unreacted protein and crosslinker.To prepare control surfaces, collagen I from rat tail (50 µg/mL in PBS) and 0.1% Poly-L-Lysine (PLL) hydrobromide solution (Polysciences, Inc. (Warrington, PA)) were incubated in wells overnight at 4 °C. All wells (including plain wells) were then rinsed three times with PBS, blocked with BSA (fraction V, 0.2% in PBS, heat inactivated at 85 °C for 10 min) to prevent non-specific binding, and rinsed three more times with PBS.
PC-12 cells (passage 6) were plated in each well at density of 15,000 cells/cm2 and differentiated in F12 Kaighn's differentiation media for 6 days. AlamarBlue® reagent (Invitrogen, Carlsbad, CA) was added directly to the media in each well at 1/10 volume and incubated at 37 °C for 13 hrs. Absorbance readings were taken at 570 nm and 600 nm. The fraction of reduced alamarBlue® reagent was calculated according to manufacturer's instructions. Statistical significance between data sets was determined using a paired, two-tailed Student's t-test.
Immunostaining
Protein films were prepared on glass coverslips as explained in the Calcein AM cell adhesion assay section (1:1 u1:RGD, u2:RGD, u3:RGD, RDG:RGD, full RGD, full RDG, and adsorbed collagen). PC-12 cells (passage 5) were plated on each film at density of 15,000 cells/cm2. After 6 days of differentiation in F12 Kaighn's differentiation media samples were rinsed twice with PBS, fixed with 4% paraformaldehyde in PBS for 15 min at room temperature, and rinsed twice more with PBS. Cells were permeabilized with 0.1% Triton X-100 (PBST, 15 min, room temperature), blocked with 10% normal goat serum (PBST, 1 h, room temperature), and incubated with rabbit monoclonal neuronal class III β-tubulin antibody (dilution factor of 1:500, Covance, Princeton, NJ, PBST containing 5% normal goat serum, room temperature, 2 hrs). Samples were then rinsed with PBST, incubated with goat anti-rabbit IgG AlexaFluor 488 (Invitrogen, Carlsbad, CA, PBST containing 5% goat serum, 1 h), and rinsed with PBST. The samples were then incubated with 1 µg/mL of 4′,6-diamidino-2-phenylindole (DAPI, Roche, Basel, Switzerland) in methanol for 15 min at 37 °C, washed with methanol, and mounted on microscope slides using Prolong Gold Antifade Reagent (Invitrogen).The fluorescently labeled cells were visualized following 6 days of differentiation with an inverted Zeiss Axiovert 200 microscope (40x objective) and digital images were captured with a CCD camera.
Neurite quantification assay
Fluorescent images were obtained as described above. A total of 94 images (field size = 0.09 mm2) were analyzed in triplicate for the RDG:RGD, full RGD, and full RDG samples and 34 images were analyzed in triplicate for the adsorbed collagen films. For each image, the total number of cells, cells with neurites greater than one cell diameter, and cells with neurites greater than two cell diameters were counted. Statistical significance between data sets was determined using a paired, two-tailed Student's t-test.Results and discussion
Material design, synthesis, and purification
The basic design of the engineered proteins and the exact amino acid sequences of each protein family member are shown in Fig. 1. Each engineered protein was designed to contain two peptide tag sequences for identification, an enterokinase cleavage site to easily remove the identification tags, and four repeats of a sequence containing both a bioactive domain and a structural domain. Five different bioactive domains were included into this basic design structure: the extended RGD cell-binding domain from fibronectin, a scrambled negative control of the cell-binding domain (RDG), and three uPA protease cleavage sites (u1, u2, u3). The RGD domain of fibronectin was selected since it has been shown previously to promote neurite attachment due to interaction with integrins displayed on the surfaces of neurites.41 The protease sites were chosen from previous work that utilized a combinatorial approach to detect the cleavage rates of specific sequences by uPA (four amino acids in length, shown in italics in Fig. 1).49 Therefore, proteins u1, u2, u3, RGD, and RDG are nearly identical and have a modest ∼3% difference in total amino acid sequence. Both the cell-binding domains and the protease cleavage domains were surrounded by flanking amino acid sequences derived from the extended fibronectin RGD domain in order to promote bioactivity within the protein polymer by facilitating conformational flexibility and potentially preserving the secondary structure of the peptide.38The structural domain included in each protein mimics the repeated VPGVG sequence found naturally in elastin, a fibrous protein present in connective tissue that is known to provide both elasticity and resilience.37 Elastin-like polypeptides have been previously shown to be biocompatible, to express well in bacterial fermentation, and to be easily and inexpensively purified using a simple thermal cycling technique.39,50 In addition to providing our scaffolds with elasticity and resilience, the elastin-like sequences provide a relatively bioinert backbone for the presentation of bioactive peptides.39
Bacterial expressions of these polymers resulted in un-optimized protein yields that ranged from 25 to 50 mg/L. Amino acid analysis of the purified protein products confirmed that the sequences were within 2% of that predicted from the primary sequence (see supplemental data). The proteins were purified using a simple thermal cycling technique made possible by the thermodynamic properties of the elastin-like sequences.35,50 The hydrophobic elastin-like sequences cause the proteins to exhibit lower critical solution temperature (LCST) behavior.51 Below the LCST, the proteins are readily soluble in water while above the LCST, the proteins aggregate to form a protein-rich coacervate that can be separated by centrifugation. Based on previous work relating specific elastin-like sequences to LCST behavior, the elastin-like sequence VPGIG was chosen to facilitate purification by thermal cycling between 4 °C and 37 °C, temperatures commonly found in a molecular biology laboratory.51 The LCST values for each protein were evaluated and found to be between 31 °C and 34 °C (see supplemental data). These LCST values supported the use of 4 °C for solubilization of the protein and 37 °C for precipitation of the protein in the thermal cycling purification technique.
Controlling scaffold mechanical properties
Lysine residues were incorporated within the elastin-like sequences as sites for polymer crosslinking using bifunctional, amine-specific, chemical crosslinkers.35 By locating the lysine residues within the elastin-like domains, the bioactive domains remain undisturbed during crosslinking and, therefore, allow the elastic modulus to be tuned independently from protein bioactivity and biodegradation. With similar reaction conditions and stoichiometries, the three protease-sensitive proteins were shown to form crosslinked scaffolds that possess similar compressive elastic moduli, confirming that the content of the bioactive sequence does not significantly affect protein crosslinking (see supplemental information). The compressive elastic modulus can be directly controlled by adjusting the location and frequency of lysine residues within the protein polymers or by altering the stoichiometry of crosslinker reactive sites to primary amines in the crosslinking reaction.35,52 Variation of the reaction stoichiometry (NHS esters: primary amines) from 0.75:1 to 1.5:1 for bulk u1 scaffolds was shown to result in a 39% increase in the compressive elastic modulus of the resultant material (Fig. 2). As shown in Table 1, our materials currently possess compressive elastic moduli within the range of 46 to 63 kPa. By modeling the scaffolds as ideal elastic networks, the crosslinking reactions are approximated to have reacted 10% of the primary amines using 0.75:1 stoichiometry and 14% of the primary amines using 1.5:1 stoichiometry.| Crosslinking Stoichiometric Ratio | Water Content (%) | Volumetric Swelling Ratio (Q) | Compressive Elastic Modulus (kPa) | Mc × 1000 (g/mol)a | Reacted Amines (%) |
|---|---|---|---|---|---|
| a Complete amine consumption yields an approximate Mc of 2,700 g/mol. | |||||
| 0.75:1 | 87.2 ± 0.1 | 10.2 ± 0.1 | 45.7 ± 7.2 | 26.8 ± 4.2 | 10.2 ± 1.6 |
| 1.5:1 | 85.4 ± 0.3 | 8.9 ± 0.2 | 63.3 ± 3.6 | 19.4 ± 1.1 | 14.1 ± 0.8 |
 | ||
| Fig. 2 Compressive stress versus strain curves for u1 protein crosslinked using two different crosslinking reaction stoichiometries. | ||
Tunable mechanical properties as evident in our materials are hypothesized to be an important characteristic in the design of a material implant. It is conjectured that matching the mechanical properties of a material to that of surrounding native tissue more closely mimics the natural cellular environment and may encourage cell movement across the implant-native tissue boundary.13,53–55 Furthermore, recent experimental evidence indicates that the mechanical strength of a material can directly affect cell growth and differentiation.22–25 There is some disagreement in the literature concerning the appropriate elastic modulus value which effectively imitates spinal cord tissue; the most common values reported range from approximately 3 kPa to 300 kPa.56,57 Our crosslinked material presently exhibits an elastic modulus within the range measured for spinal cord tissue.
Another key attribute of our crosslinked protein scaffolds is their highly swollen, hydrogel structure. Highly swollen networks allow for diffusion of nutrients and growth factors and provide open space for the migration of cells through a material implant.58 The water content of the swollen u1 materials was measured to be 85–87% and the volumetric swelling ratios of the materials were calculated to be from 8 to10 (Table 1). The water content of these materials are comparable to other nerve regeneration scaffolds constructed from PHEMA (85% water content) and are slightly less swollen than degradable PEG hydrogels previously used for neural applications (Q ∼12–16).13,55
Tailored scaffold enzymatic degradation
Despite the highly swollen nature of these protein materials, we hypothesize that they may still be obstructive to nerve cells attempting to regenerate through the polymer network. This hypothesis is based on previous work that showed neurite extension was enhanced in faster degrading PEG polymer networks that possessed swelling ratios greater than our materials (Q>10).13 Therefore, we may be able to tune neurite extension by including protease cleavage sequences within the protein structure to provide a mechanism for neurons to locally degrade and remodel the material as they extend neurites. To alter the rate of proteolytic degradation, three uPA cleavage sequences with altered substrate specificity were designed into the scaffold, with u1 predicted to degrade the fastest, followed by u2 and then u3.The degradation properties of these proteins were first characterized without chemical crosslinking in the presence of uPA under physiological conditions. The high enzyme concentration (528 U/mL) used in these reactions was not chosen to mimic in vivo concentrations of uPA, but was instead selected to be similar in range to previous uPA kinetics experiments in order to create more manageable experimental time points.59 Gel electrophoresis of the degradation fragments present at a range of time points for a representative u1 degradation reaction is shown in Fig. 3. As shown in this gel, the full protein degrades to smaller fragments throughout the course of the reaction and predominantly results in the formation of one final fragment, approximately 10 kDa in weight (circled). This band was excised from the gel and sequenced to determine the specificity of the degradation reaction. The band was found to possess an N-terminal sequence of SASPASSA, which is present directly after the predicted protease cut site, arginine, (Fig. 1), thus confirming the degradation reaction specificity.
 | ||
| Fig. 3 Representative SDS-PAGE gel of u1 protein fragments generated by degradation with 528 U/mL uPA enzyme (circled band was analyzed by N-terminal sequencing). | ||
A more thorough analysis of the multiple degradation bands on the gel further supports the reaction specificity. Evident in Fig. 3 is the presence of very distinct protein degradation bands, as opposed to blurred bands, indicating highly specific cuts. The full u1 protein (37.7 kDa) contains four engineered cut sites (Fig. 1) allowing for the possible formation of 11 different sizes of specifically degraded fragments (see supplemental data for detailed mapping of gel fragments). The presence of less intense minor bands on the gel are likely due to uPA cleavage at lysine residues in the protein, which has been previously shown to occur at a much slower rate than cleavage at the engineered arginine cut sites.59 Many of these lysine residues will be consumed after chemically crosslinking the proteins, thus limiting the occurrence of these minor reactions.
Densitometry analysis was performed on SDS-PAGE gels run in triplicate to directly compare the degradation rates of u1, u2, and u3. The u1 protein degraded approximately 200-times faster than u3, and the u2 protein degraded approximately 11-times faster than the u3 protein, Fig. 4. The proteins were shown to have the following half-lives: 0.1 hours for u1, 1.1 hours for u2, and 20.6 hours for u3, when exposed to high uPA concentrations (5 U/mL). The results of this analysis were further corroborated using a colorimetric reaction to detect the newly created amine groups upon each peptide bond cleavage event for the two proteins with appreciable degradation: u1 and u2. Engineered protein u1 was shown to produce free amines at a significantly higher rate than u2, corresponding to a faster degradation rate (Fig. 5). The uPA enzyme is known to follow standard Michaelis-Menten enzyme kinetics, and plots of cleavage rate versus substrate protein concentration fit well to the Michaelis-Menten model. For the u1 protein, kcat and Km were determined to be 14.5 ± 2.2 s−1 and 327.7 ± 24.9 µM, respectively. We were unable to determine exact values of kcat and Km for the u2 protein due to significant fitting error, likely resulting from high viscosity (and hence poor mixing) of protein solutions above the predicted Km. However, the observed maximum reaction velocity of u1 was approximately 10-times greater than u2. Taken together, these results demonstrate that molecular-level design of the engineered proteins can vary the degradation rate over two orders of magnitude and substantiate the transfer of bioactivity from a short peptide sequence to the full-length protein polymer.
 | ||
| Fig. 4 Relative degradation rates of uncrosslinked u1 and u2 (inset of u3) generated by SDS-PAGE densitometry analysis of the degradation fragments produced over time upon exposure to 528 U/mL uPA enzyme. | ||
 | ||
| Fig. 5 Rate of primary amine production from degradation of uncrosslinked u1 and u2 protein with 270 U/µL of uPA enzyme. | ||
To confirm that these differences in protein degradation would result in altered degradation of the bulk scaffold, each engineered protein was chemically crosslinked to form a three-dimensional sheet (1:1 NHS esters: primary amines crosslinking stoichiometry). Each bulk scaffold was exposed to 300 U/mL uPA in buffer at room temperature. This high enzyme concentration was chosen to create manageable timepoints to accurately measure the compressive elastic modulus of each sample. The degradation of the materials was evaluated by monitoring the change in compressive elastic modulus over time. Crosslinked scaffolds displayed tunable degradation properties similar to the uncrosslinked protein polymers; the u1 protein fully degraded at around 24 hours, the u2 protein fully degraded at around 164 hours, and the u3 protein showed no detectable degradation after 164 hours (Fig. 6). These results demonstrate that by changing only 3% of the amino acid side chains in the engineered proteins, scaffolds with similar initial mechanical properties and dramatically altered degradation rates can be created.
 | ||
| Fig. 6 Change in compressive elastic modulus due to degradation of crosslinked u1, u2, and u3 scaffolds with 300 U/mL uPA enzyme. | ||
Tuning PC-12 neuronal-like cell adhesivity
In vitro cell culture studies using the model PC-12 neuronal-like cell line were conducted on crosslinked scaffolds (1:1 NHS esters: primary amines crosslinking stoichiometry) of the engineered proteins to assess cell compatibility, adhesion, and differentiation. PC-12 cells are a clonal line of rat pheochromocytoma cells that extend neuronal-like processes when exposed to NGF and are known to secrete uPA from the tips of these processes.43 Cell adhesion and viability were assessed in a quantitative manner for PC-12 cells grown on the engineered protein scaffolds in both growth (i.e., non-differentiation) and differentiation media. Crosslinked scaffolds were created using the individual engineered proteins alone and in various combinations. By crosslinking multiple engineered proteins together into a single, homogeneous scaffold, the degradation rate and cell adhesivity of the scaffold can be independently tuned. As a negative control for cell adhesion, the RGD cell-binding domain of fibronectin was replaced with the scrambled RDG domain, which is a well-known negative control for cell adhesion.60 Therefore, this negative control allows side-by-side comparison of cell adhesion and differentiation on scaffolds that have exactly identical amino acid content, identical mechanical properties, and identical degradation properties in order to determine if cell adhesion is specifically a consequence of the RGD domain.PC-12 cell adhesion was evaluated after 6 days of culture in growth medium without differentiation cues on the crosslinked protein scaffolds using calecin AM staining (Fig. 7, see supplemental data for detailed statistical comparison). Collagen, a material commonly used to promote PC-12 adhesion,61 was used as a positive control, while the negative control was bovine serum albumin (BSA), a common blocking agent to prevent adhesion. Cell adhesion on the RDG scaffold and BSA negative controls was significantly less than on all other substrates. This result confirms that the elastin backbone represents a relatively inert background upon which biofunctional peptides can be presented. Cell adhesion on scaffolds with a 1:1 ratio of RDG:RGD proteins is significantly greater than adhesion on RDG scaffolds, confirming that cell adhesion is due to specific cellular interactions with the RGD peptide sequence. All scaffolds comprised of 50% RGD protein (with remaining 50% of u1, u2, u3, or RDG), showed similar cell adhesivity. This result implies that the protease degradation sequences do not interfere with nor contribute to cell adhesion; thereby allowing independent tuning of cell adhesivity and scaffold degradation.
 | ||
| Fig. 7 Comparison of PC-12 cell adhesion after 6 days of growth without differentiation medium on various substrates. *BSA and RDG are statistically different (p < 0.05) from all other substrates. | ||
Next, PC-12 cell adhesion was evaluated after 6 days of culture in differentiation medium (in the presence of 50 ng/mL NGF) by monitoring the reduction of the alamarBlue® reagent (Fig. 8, see supplemental data for detailed statistical comparison). An increase in reagent reduction correlates to an increase in metabolic intermediates (e.g. NADH, NADPH, and FADH) produced by viable cells; therefore reagent reduction is commonly used as a quantitative measure of the number of cells present.62 An adsorbed poly-L-lysine (PLL) coating is included as an additional positive control. PLL is a commonly used substrate that promotes neuronal adhesion through electrostatic interactions.61 Most evident from this assay is the dramatic decrease in cells present when RGD is replaced by RDG (u1:RGD vs. u1:RDG (p = 0.027), u2:RGD vs. u2:RDG (p = 0.028), u3:RGD vs. u3:RDG (p = 0.067), and RDG:RGD vs. RDG (p = 0.002)). Also, the number of cells present is similar in u1:RGD, u2:RGD, u3:RGD, and RDG:RGD, again confirming that scaffold degradation can be tuned independently from cell adhesivity.
 | ||
| Fig. 8 Comparison of PC-12 cell adhesion after 6 days of differentiation on various substrates. *Substrates are statistically different, p < 0.05. | ||
In addition, these results suggest that the degree of cell adhesivity can be tuned by controlling the density of RGD cell-binding domains within the crosslinked scaffold. Scaffolds comprised entirely of the RGD protein are estimated to contain ∼ 9,300 RGD binding sites/µm2, while 1:1 RDG:RGD scaffolds contain ∼ 4,650 and RDG scaffolds contain 0 RGD binding sites/µm2; assuming the scaffold density is 8.2 µg protein/mm3 (estimated from swelling data in Table 1 and the concentration of protein in the spincoated crosslinking solutions) and cell integrins can penetrate a depth of 18 nm into the scaffold.63 Comparing PC-12 cell adhesion on these three protein scaffolds, we observe that cell adhesion is directly related to RGD binding site density, Fig. 8. Therefore, by tuning the relative ratio of RGD and RDG proteins, we can tailor the cell adhesivity of the scaffold.
Tuning differentiation and neurite outgrowth
The ability of the engineered protein scaffolds to support PC-12 cell differentiation into a neuronal phenotype was analyzed using immunocytochemistry following 6 days of culture in differentiation medium (in the presence of 50 ng/mL NGF), Fig. 9. PC-12 cells were observed to adhere and extend neuritic-like processes on all of the engineered protein scaffolds (u1:RGD, u2:RGD, u3:RGD, RDG:RGD, and RGD) containing the RGD cell-binding site. The neuronal-like cell morphology and positive staining for neuron-specific β-tubulin indicates that these scaffolds support cell proliferation, differentiation, and neurite outgrowth and show no evidence of cytotoxicity. Little neurite extension was observed on the RDG scaffold or the adsorbed BSA negative controls, confirming that the RGD peptide must be present to support neurite outgrowth. Cell morphology on the u1:RGD, u2:RGD, and u3:RGD scaffolds appear similar, suggesting that the protease degradation sequences do not interfere with nor contribute to neurite outgrowth, allowing scaffold degradation to be tuned independent of neurite outgrowth. Cells grown on scaffolds containing 100% RGD protein (∼ 9,300 RGD binding sites/µm2 of RGD binding sites) exhibit a similar morphology to that observed on adsorbed collagen positive control surfaces; while cells grown on scaffolds containing 50% RGD protein (∼ 4,650 RGD binding sites/µm2) exhibit fewer neurites with less developed branching. | ||
| Fig. 9 Representative fluorescent images of PC-12 cells differentiated for 6 days on a) u1:RGD b) u2:RGD c) u3:RGD d) RDG:RGD e) RGD f) Collagen g) RDG h) BSA; nuclei stained with DAPI in blue, cytoplasm stained with neuron-specific β-tubulin in green, scale bar = 50 µm. | ||
To quantitatively assess the morphological differences caused by RGD binding site density, 94 images of each engineered scaffold were analyzed in triplicate, (Fig. 10, see supplemental data for detailed statistical comparison). By increasing the total amount of RGD present in the film, the number of adherent cells, the percentage of cells possessing neurites greater than one cell diameter in length, and the percentage of cells possessing neurites greater than two cell diameters in length were all found to increase. The RDG film was statistically less adherent to differentiated cells than all other scaffolds (Fig. 10a). The percentage of cells with neurites greater than one cell diameter (Fig. 10b) and the percentage of cells with neurites greater than two cell diameters (Fig. 10c) were statistically different on scaffolds with 0; 4,650; and 9,300 RGD binding sites/µm2. The scaffold comprised solely of RGD protein differentiated fewer cells than the positive collagen control, indicating that further optimization of the cell adhesion ligand density and the addition of other adhesion sequences should be investigated to further enhance the ability of these designed materials to support cell adhesion, neuronal differentiation, and neurite outgrowth.27,41
 | ||
| Fig. 10 Comparison of PC-12 cells after 6 days of differentiation on RDG, 1:1 RDG:RGD, RGD, and collagen films in terms of a) cells/mm2 b) percentage of cells with neurites greater than one cell diameter and c) percentage of cells with neurites greater than two cell diameters. *Substrate is statistically different from all other substrates, p < 0.05. | ||
Conclusions
The overall goal of this research is to use synthetic proteins to fabricate a regenerative scaffold that can be systematically tailored to obtain the optimal stimuli required for spinal cord nerve regeneration. While many biomaterials have been developed to carefully control a single material parameter for systematic cell-scaffold interaction studies, few, if any, biomaterials have been designed to allow independent, simultaneous tuning of initial elasticity, degradation rate, and cell adhesivity. Because all three variables are known to impact neurite outgrowth, it is critical to engineer new scaffolds for CNS nerve grafts that allow systematic tailoring of each biomaterial parameter.The tunable cell adhesion, neuronal differentiation, and neurite outgrowth properties displayed by our engineered proteins encourage further use of these scaffolds to study the optimal initial mechanics, degradation rate, and ligand content to encourage nerve regeneration. While several groups are investigating the effects of elastic modulus and ligand density on neurite outgrowth,24,25,27,28 at present the optimal degradation rate to promote neurite outgrowth is largely unknown. Furthermore, the potential cross-talk between these multiple points of cell-scaffold interaction are only just beginning to be explored.30,64 These types of important studies require the design of biomaterials that allow the systematic tuning of specific material parameters without impacting other material properties.
In specific, by using the precise protein synthesis machinery present in bacteria, we have designed a family of biomaterials that mimics cell-adhesive and cell-responsive properties of the regenerative nerve environment. Here we report the crosslinking of these proteins into scaffolds with tailored initial elastic moduli. We also report the tunable biodegradation of these engineered scaffolds in response to enzymatic cleavage by uPA, a protease known to be secreted by the tips of growing neurites. By exploiting the sensitivity of uPA substrate specificity to slight changes in the neighboring amino acid sequence, we synthesized three proteins with highly variable degradation rates. The uncrosslinked proteins were shown to exhibit a greater than 200-fold change in protease degradation half-life as a result of changing only 3% of the total amino acid content. This same change in sequence translated into scaffolds with degradation lifetimes that varied from full degradation after one day of constant uPA exposure to no detected degradation after one full week of uPA exposure.
Through the design of three different scaffolds (u1, u2, and u3) with identical cell adhesion properties, identical initial elastic moduli, and significantly different degradation rates, we aim to provide a mechanism to study the optimal degradation rate for neurite outgrowth. The three uPA degradable proteins can be further combined together at different ratios to create an even larger set of materials to be studied for regenerative properties. Previous work has encouraged the development of this strategy by showing that cells attempting to grow through quicker degrading materials often grow faster and extend longer projections.13,14,19,21 Despite these advancements, we hypothesize that in order to maximize the benefit of a supportive scaffold, the degradation rate must closely match the cell regeneration rate and the site of degradation must be highly localized to prevent collapse of the bulk scaffold. Therefore, we have specifically targeted a protease secreted locally by the tips of growing neurites, which are the sites of regeneration in neurons, in order to more closely mimic what is believed to occur in the natural ECM environment.42 Future work will evaluate the rates of uPA secretion, neurite extension of neuronal cells in two-dimensional and three-dimensional scaffolds of varying mechanical properties and degradation rates, and the immunogenicity of implanted scaffolds.
A composite scaffold of the uPA-sensitive engineered proteins and an RGD engineered protein was shown to support cell attachment, neuronal differentiation, and neurite extension of a PC-12 neuronal-like cell line. The total amount of RGD present in the proteins was found to directly control both cell adhesion and neurite outgrowth. Taken together, these data demonstrate that the initial elastic moduli, degradation rate, and cell adhesivity of the scaffolds can each be systematically and independently tuned. These engineered proteins will be used to identify the combination of optimal material properties that promotes neurite outgrowth. Ultimately, knowledge obtained from these proteins may be applied to the construction of a biodegradable material that delivers a combination of beneficial stimuli at the site of nerve injury. Together with a combined approach involving the delivery of cells and growth factors, these multi-component protein polymers offer a new approach to spinal cord repair.
Acknowledgements
The authors acknowledge funding support from the National Academies Keck Futures Initiative, the John and Ulla deLarios Scholar Fund, and the Hellman Faculty Scholar Fund. We thank the Stanford Center on Polymer Interfaces and Macromolecular Assemblies for the use of equipment.References
- C. M. Galtrey, R. A. Asher, F. Nothias and J. W. Fawcett, Brain, 2007, 130, 926–939.
- M. G. Fehlings and R. G. Perrin, Spine, 2006, 31, S28–S35 CrossRef.
- C. E. Schmidt and J. B. Leach, Annual Review of Biomedical Engineering, 2003, 5, 293–347 Search PubMed.
- P. M. Richardson, U. M. McGuinness and A. J. Aguayo, Nature, 1980, 284, 264–265 CrossRef CAS.
- H. Nomura, C. H. Tator and M. S. Shoichet, Journal of Neurotrauma, 2006, 23, 496–507 Search PubMed.
- B. Schlosshauer, L. Dreesmann, H. E. Schaller and N. Sinis, Neurosurgery, 2006, 59, 740–747 Search PubMed.
- Y. C. Huang and Y. Y. Huang, Artificial Organs, 2006, 30, 514–522 Search PubMed.
- J. Ijkema-Paassen, K. Jansen, A. Gramsbergen and M. F. Meek, Biomaterials, 2004, 25, 1583–1592 CrossRef CAS.
- J. S. Belkas, M. S. Shoichet and R. Midha, Neurological Research, 2004, 26, 151–160 Search PubMed.
- R. C. Young, M. Wiberg and G. Terenghi, British Journal of Plastic Surgery, 2002, 55, 235–240 Search PubMed.
- A. S. Sawhney, C. P. Pathak and J. A. Hubbell, Macromolecules, 1993, 26, 581–587 CrossRef CAS.
- A. T. Metters, K. S. Anseth and C. N. Bowman, Polymer, 2000, 41, 3993–4004 CrossRef CAS.
- M. J. Mahoney and K. S. Anseth, Biomaterials, 2006, 27, 2265–2274 CrossRef CAS.
- R. Pittier, F. Sauthier, J. A. Hubbell and H. Hall, Journal of Neurobiology, 2005, 63, 1–14 Search PubMed.
- T. Freier, H. S. Koh, K. Kazazian and M. S. Shoichet, Biomaterials, 2005, 26, 5872–5878 CrossRef CAS.
- N. Inoue, M. Bessho, M. Furuta, T. Kojima, S. Okuda and M. Hara, Journal of Biomaterials Science, Polymer Edition, 2006, 17, 837–858 Search PubMed.
- S. G. Levesque and M. S. Shoichet, Bioconjugate Chemistry, 2007, 18, 874–885 CrossRef CAS.
- J. L. West and J. A. Hubbell, Macromolecules, 1999, 32, 241–244 CrossRef CAS.
- S. C. Rizzi, M. Ehrbar, S. Halstenberg, G. P. Raeber, H. G. Schmoekel, H. Hagenmuller, R. Muller, F. E. Weber and J. A. Hubbell, Biomacromolecules, 2006, 7, 3019–3029 CrossRef CAS.
- S. Kim, E. H. Chung, M. Gilbert and K. E. Healy, Journal of Biomedical Materials Research Part A, 2005, 75A, 73–88 Search PubMed.
- M. P. Lutolf, J. L. Lauer-Fields, H. G. Schmoekel, A. T. Metters, F. E. Weber, G. B. Fields and J. A. Hubbell, Proceedings of the National Academy of Sciences of the United States of America, 2003, 100, 5413–5418 Search PubMed.
- A. P. Balgude, X. Yu, A. Szymanski and R. V. Bellamkonda, Biomaterials, 2001, 22, 1077–1084 CrossRef CAS.
- I. Levental, P. C. Georges and P. A. Janmey, Soft Matter, 2007, 3, 299–306 RSC.
- J. B. Leach, X. Q. Brown, J. G. Jacot, P. A. DiMilla and J. Y. Wong, Journal of Neural Engineering, 2007, 4, 26–34 Search PubMed.
- L. A. Flanagan, Y. E. Ju, B. Marg, M. Osterfield and P. A. Janmey, Neuroreport, 2002, 13, 2411–2415 CrossRef.
- G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I. Stupp, Science, 2004, 303, 1352–1355 CrossRef CAS.
- V. M. Tysseling-Mattiace, V. Sahni, K. L. Niece, D. Birch, C. Czeisler, M. G. Fehlings, S. I. Stupp and J. A. Kessler, Journal of Neuroscience, 2008, 28, 3814–3823 Search PubMed.
- L. M. Y. Yu, K. Kazazian and M. S. Shoichet, Journal of Biomedical Materials Research Part A, 2007, 82A, 243–255 Search PubMed.
- P. Musoke-Zawedde and M. S. Shoichet, Biomedical Materials, 2006, 1, 162–169 CrossRef CAS.
- L. Little, K. E. Healy and D. Schaffer, Chemical Reviews, 2008, 108, 1787–1796 CrossRef CAS.
- M. E. Davis, P. C. H. Hsieh, A. J. Grodzinsky and R. T. Lee, Circulation Research, 2005, 97, 8–15 Search PubMed.
- J. S. Belkas, C. A. Munro, M. S. Shoichet, M. Johnston and R. Midha, Biomaterials, 2005, 26, 1741–1749 CrossRef CAS.
- M. Oudega, S. E. Gautier, P. Chapon, M. Fragoso, M. L. Bates, J. M. Parel and M. B. Bunge, Biomaterials, 2001, 22, 1125–1136 CrossRef CAS.
- I. Yamaguchi, S. Itoh, M. Suzuki, A. Osaka and J. Tanaka, Biomaterials, 2003, 24, 3285–3292 CrossRef CAS.
- K. Di Zio and D. A. Tirrell, Macromolecules, 2003, 36, 1553–1558 CrossRef.
- M. K. McHale, L. A. Setton and A. Chilkoti, Tissue Engineering, 2005, 11, 1768–1779 Search PubMed.
- A. Nicol, D. C. Gowda and D. W. Urry, Journal of Biomedical Materials Research, 1992, 26, 393–413 Search PubMed.
- S. C. Heilshorn, K. A. DiZio, E. R. Welsh and D. A. Tirrell, Biomaterials, 2003, 24, 4245–4252 CrossRef.
- S. C. Heilshorn, J. C. Liu and D. A. Tirrell, Biomacromolecules, 2005, 6, 318–323 CrossRef CAS.
- D. W. Urry, T. M. Parker, M. C. Reid and D. C. Gowda, Journal of Bioactive and Compatible Polymers, 1991, 6, 263–282 Search PubMed.
- S. Meiners and M. L. T. Mercado, Molecular Neurobiology, 2003, 27, 177–195 Search PubMed.
- N. W. Seeds, L. B. Siconolfi and S. P. Haffke, Cell and Tissue Research, 1997, 290, 367–370 Search PubMed.
- R. N. Pittman, J. K. Ivins and H. M. Buettner, The Journal of Neuroscience, 1989, 9, 4269–4286 Search PubMed.
- M. E. Schwab, Science, 2002, 295, 1029–1031 CrossRef CAS.
- H. M. Geller and J. W. Fawcett, Experimental Neurology, 2002, 174, 125–136 Search PubMed.
- L. N. Novikova, L. N. Novikov and J. O. Kellerth, Current Opinion in Neurology, 2003, 16, 711–715 Search PubMed.
- H. Fischer, I. Polikarpov and A. F. Craievich, Protein Science, 2004, 13, 2825–2828 CrossRef CAS.
- Y. C. Fung, Biomechanics: Mechanical Properties of Living Tissues, Springer-Verlag, New York, 1993 Search PubMed.
- J. L. Harris, B. J. Backes, F. Leonetti, S. Mahrus, J. A. Ellman and C. S. Craik, Proceedings of the National Academy of Sciences of the United States of America, 2000, 97, 7754–7759 Search PubMed.
- D. E. Meyer and A. Chilkoti, Nature Biotechnology, 1999, 17, 1112–1115 CrossRef CAS.
- D. W. Urry, D. C. Gowda, T. M. Parker, C. H. Luan, M. C. Reid, C. M. Harris, A. Pattanaik and R. D. Harris, Biopolymers, 1992, 32, 1243–1250 CrossRef CAS.
- P. J. Nowatzki and D. A. Tirrell, Biomaterials, 2004, 25, 1261–1267 CrossRef CAS.
- P. D. Dalton, L. Flynn and M. S. Shoichet, Biomaterials, 2002, 23, 3843–3851 CrossRef CAS.
- S. Woerly, E. Pinet, L. de Robertis, D. Van Diep and M. Bousmina, Biomaterials, 2001, 22, 1095–1111 CrossRef CAS.
- A. Bakshi, O. Fisher, T. Dagci, B. T. Himes, I. Fischer and A. Lowman, Journal of Neurosurgery-Spine, 2004, 3, 322–329 Search PubMed.
- R. J. Oakland, R. M. Hall, R. K. Wilcox and D. C. Barton, Proceedings of the Institution of Mechanical Engineers Part H-Journal of Engineering in Medicine, 2006, 220, 489–492 Search PubMed.
- H. Ozawa, T. Matsumoto, T. Ohashi, M. Sato and S. Kokubun, Journal of Neurosurgery, 2001, 95, 221–224 Search PubMed.
- N. A. Peppas, Y. Huang, M. Torres-Lugo, J. H. Ward and J. Zhang, Annual Review of Biomedical Engineering, 2000, 2, 9–29 Search PubMed.
- S. H. Ke, G. S. Coombs, K. Tachias, D. R. Corey and E. L. Madison, Journal of Biological Chemistry, 1997, 272, 20456–20462 Search PubMed.
- J. C. Liu, S. C. Heilshorn and D. A. Tirrell, Biomacromolecules, 2004, 5, 497–504 CrossRef CAS.
- G. Banker, L. A. Goslin, ed., Culturing Nerve Cells, 2nd edn., MIT Press, Cambridge, MA, 1998 Search PubMed.
- H. Gazzano-Santoro, A. Chen, B. Casto, H. Chu, E. Gilkerson, V. Mukku, E. Canova-Davis and C. Kotts, Journal of Pharmaceutical and Biomedical Analysis, 1999, 21, 945–959 Search PubMed.
- J. P. Xiong, T. Stehle, B. Diefenbach, R. G. Zhang, R. Dunker, D. L. Scott, A. Joachimiak, S. L. Goodman and M. A. Arnaout, Science, 2001, 294, 339–345 CrossRef CAS.
- M. H. Zaman, P. Matsudaira and D. A. Lauffenburger, Annals of Biomedical Engineering, 2007, 35, 91–100 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Plasmid cloning procedure, amino acid anlaysis of purified protein, lower critical solution temperatures of proteins, analysis of u1 protein degradation fragments, representative full stress-strain curve from compression testing, comparison of protein compressive elastic moduli with similar crosslinking conditions, and Student's t-test p-values for PC-12 cell experiments. See DOI: 10.1039/b808504h |
| This journal is © The Royal Society of Chemistry 2009 |